As indicated in my first post-holiday post, my flasks mostly still have live cells – although some of the more confluent flasks also resulted in mass cell death. Interestingly, my first thawed flask of cells – with remaining cells maintained after the first passage – still has living cells after four months of continuous culture:
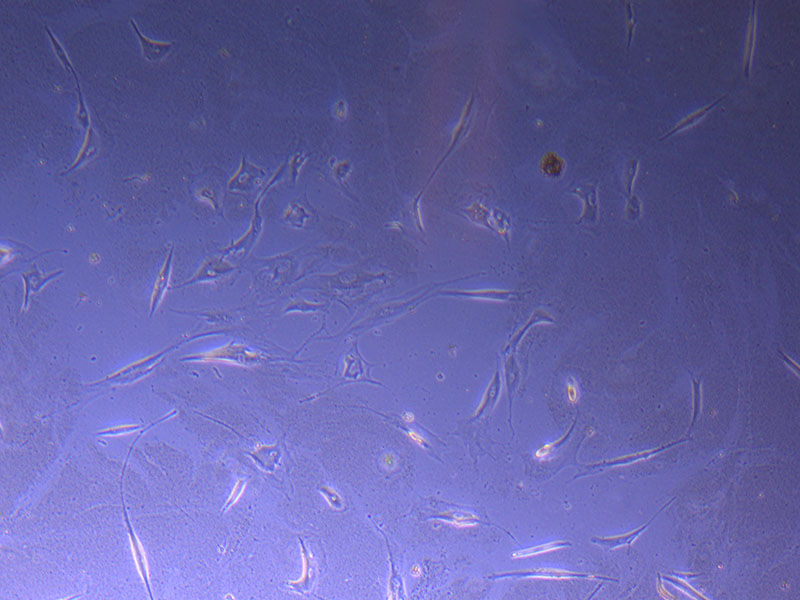 Light microscope image of PHGL TB cells at 13/1/22 showing a main cluster area of remaining cells.
Light microscope image of PHGL TB cells at 13/1/22 showing a main cluster area of remaining cells.
These cells were originally thawed and plated on 10/09/21. After passage on 21/09/21 the original flask continued to be maintained with a weekly feeding/media change regimen.
 Photograph of P1 PHGL TB flask originally plated out by Jo-Maree on 10/0921.
Photograph of P1 PHGL TB flask originally plated out by Jo-Maree on 10/0921.
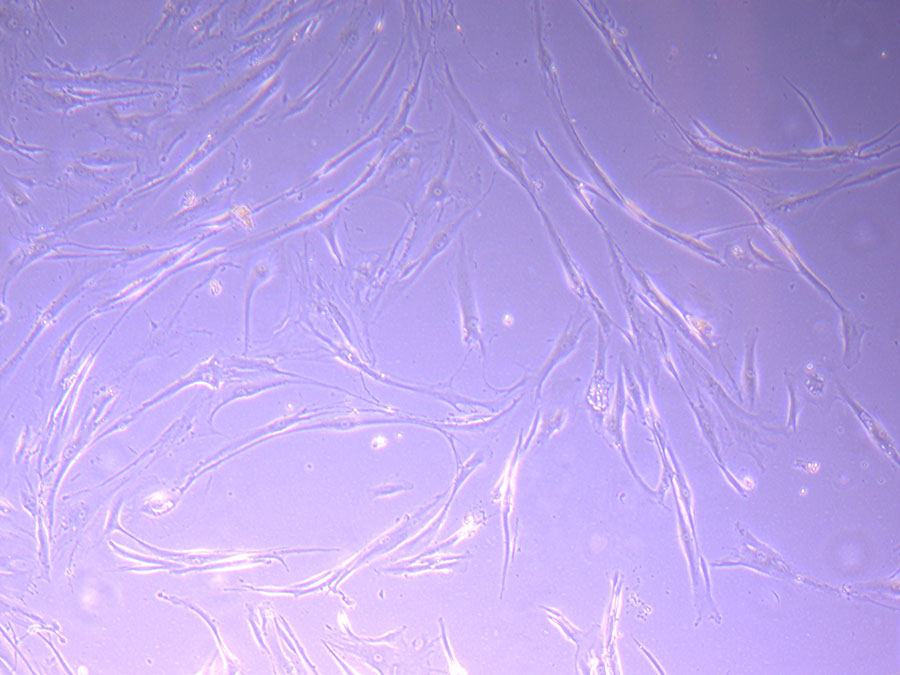 Light microscope image of P1 PHGL TB cells taken on 21/09/21 prior to passage.
Light microscope image of P1 PHGL TB cells taken on 21/09/21 prior to passage.
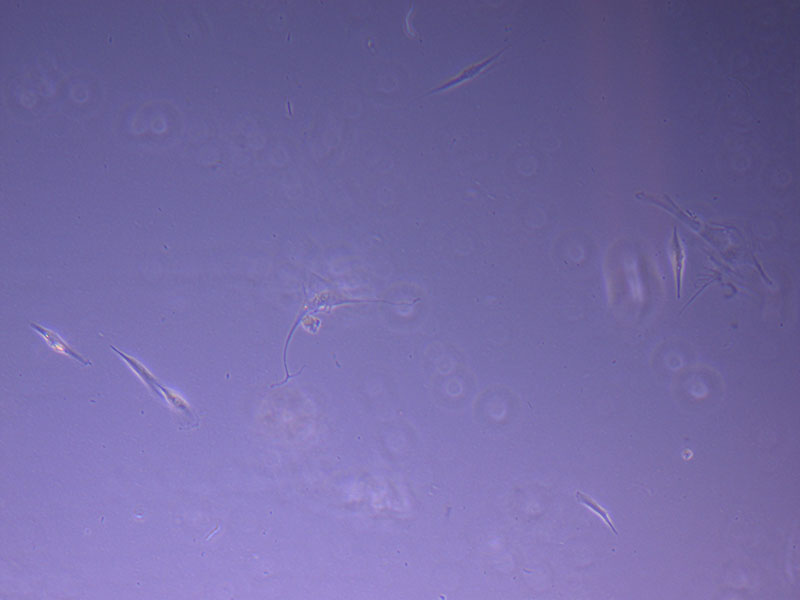 Light microscope image of P2 PHGL TB cells (in original flask) taken on 23/09/21 after cell passage on 21/09/21. A few cells remain visible in the flask.
Light microscope image of P2 PHGL TB cells (in original flask) taken on 23/09/21 after cell passage on 21/09/21. A few cells remain visible in the flask.
At present, here are only very small clusters of cells remaining:
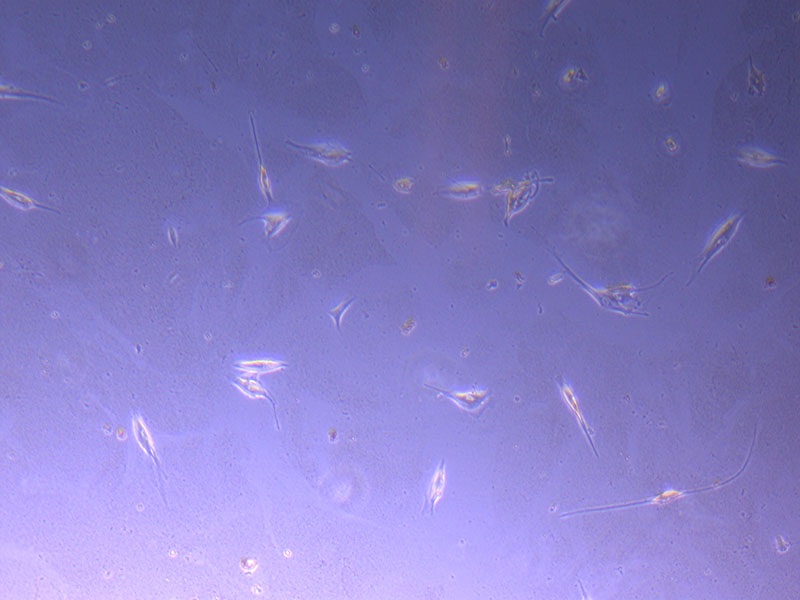 Light microscope image of P2 PHGL TB cells (in original flask) taken on 13/1/22. The image reveals a ‘sprinkle’ of cells beyond the main cluster.
Light microscope image of P2 PHGL TB cells (in original flask) taken on 13/1/22. The image reveals a ‘sprinkle’ of cells beyond the main cluster.
The rest of the flask is filled with the ghostly trails of cellular movement:
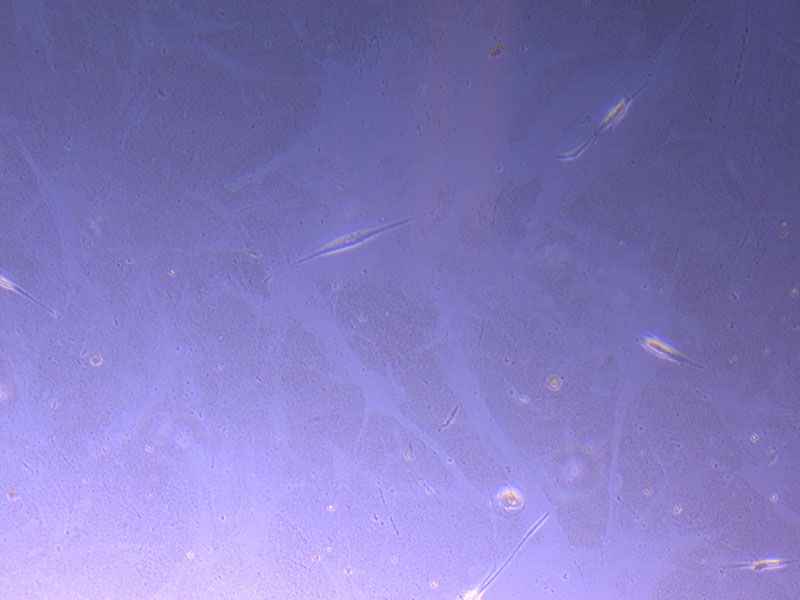
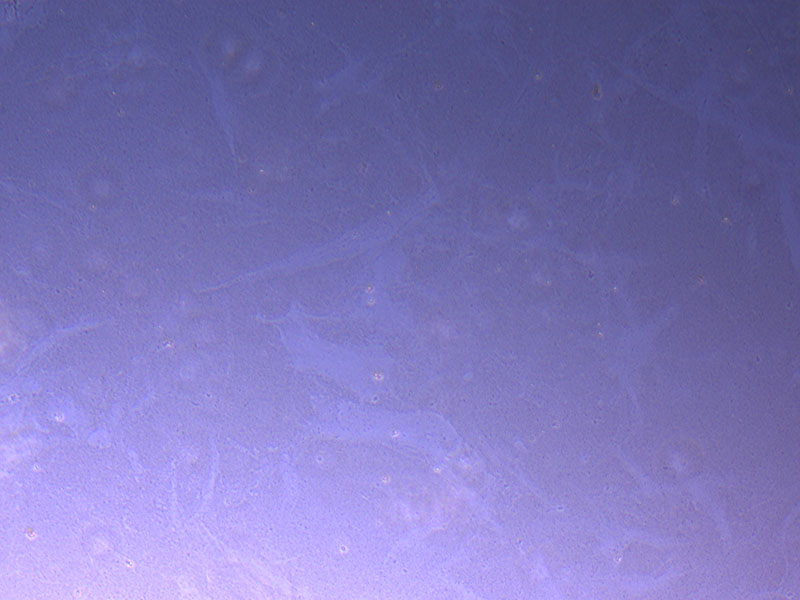 Light microscope images of P2 PHGL TB cells (in original flask) taken on 13/1/22. The image reveals trails of cellular movement and existence.
Light microscope images of P2 PHGL TB cells (in original flask) taken on 13/1/22. The image reveals trails of cellular movement and existence.
Since this is pretty consistent with prolonged culture, I am curious to show Jo-Maree to determine what the ‘residue’ is. It is quite poetic to consider the way in which traces remain of different movements and interactions. I have decided to continue to maintain the flask until there are no more viable cells, so it will be interesting to see how these traces evolve.
 Light microscope image of cut glass dish with media containing dead cell debris and evidence of a small number of surviving cells.
Light microscope image of cut glass dish with media containing dead cell debris and evidence of a small number of surviving cells. 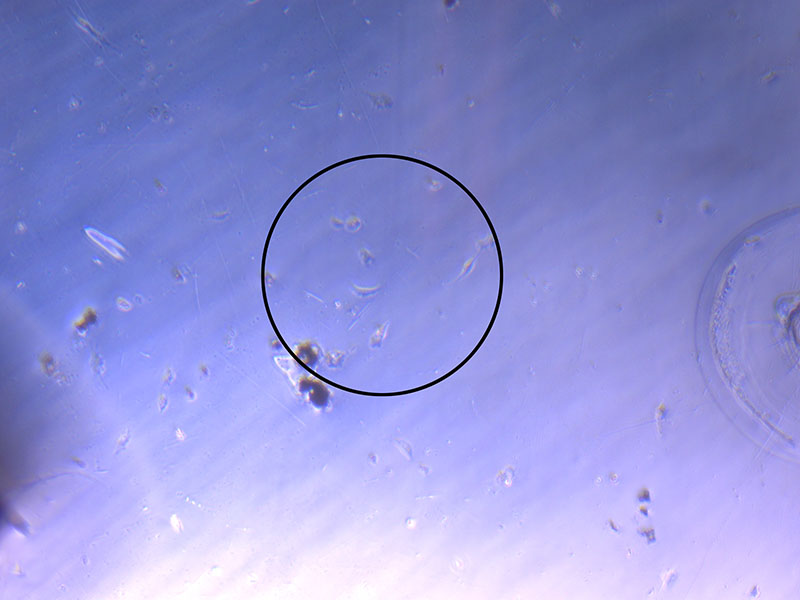
 Light microscope image of cut glass dish in PBS showing further evidence of a small number of surviving cells.
Light microscope image of cut glass dish in PBS showing further evidence of a small number of surviving cells.  Light microscope image of cut glass dish in PBS with likely cells circled. There are a few additional potential cells visible, but I have only circled the most obvious.
Light microscope image of cut glass dish in PBS with likely cells circled. There are a few additional potential cells visible, but I have only circled the most obvious. 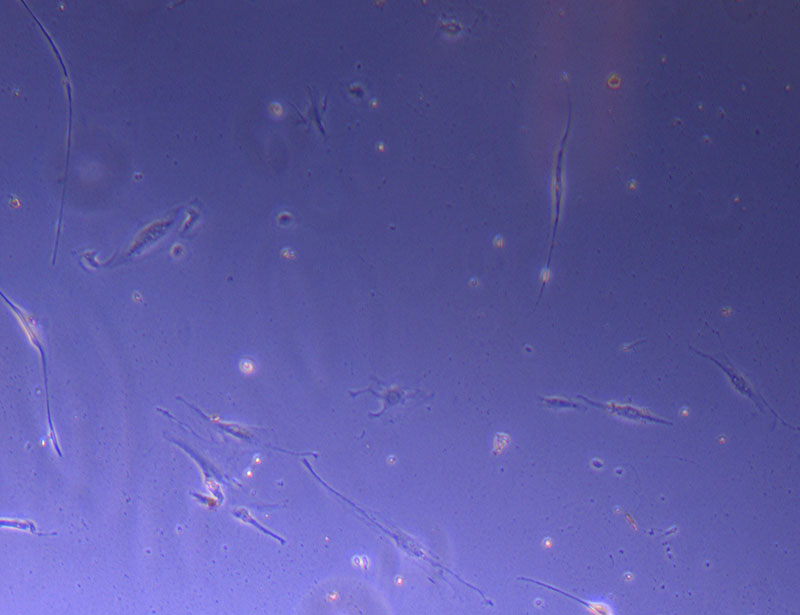 Light microscope image of Petri dish with fresh complete media with live cells.
Light microscope image of Petri dish with fresh complete media with live cells. 
 Simple graphic interface with presets ready to complete fluorescent microscopy.
Simple graphic interface with presets ready to complete fluorescent microscopy. 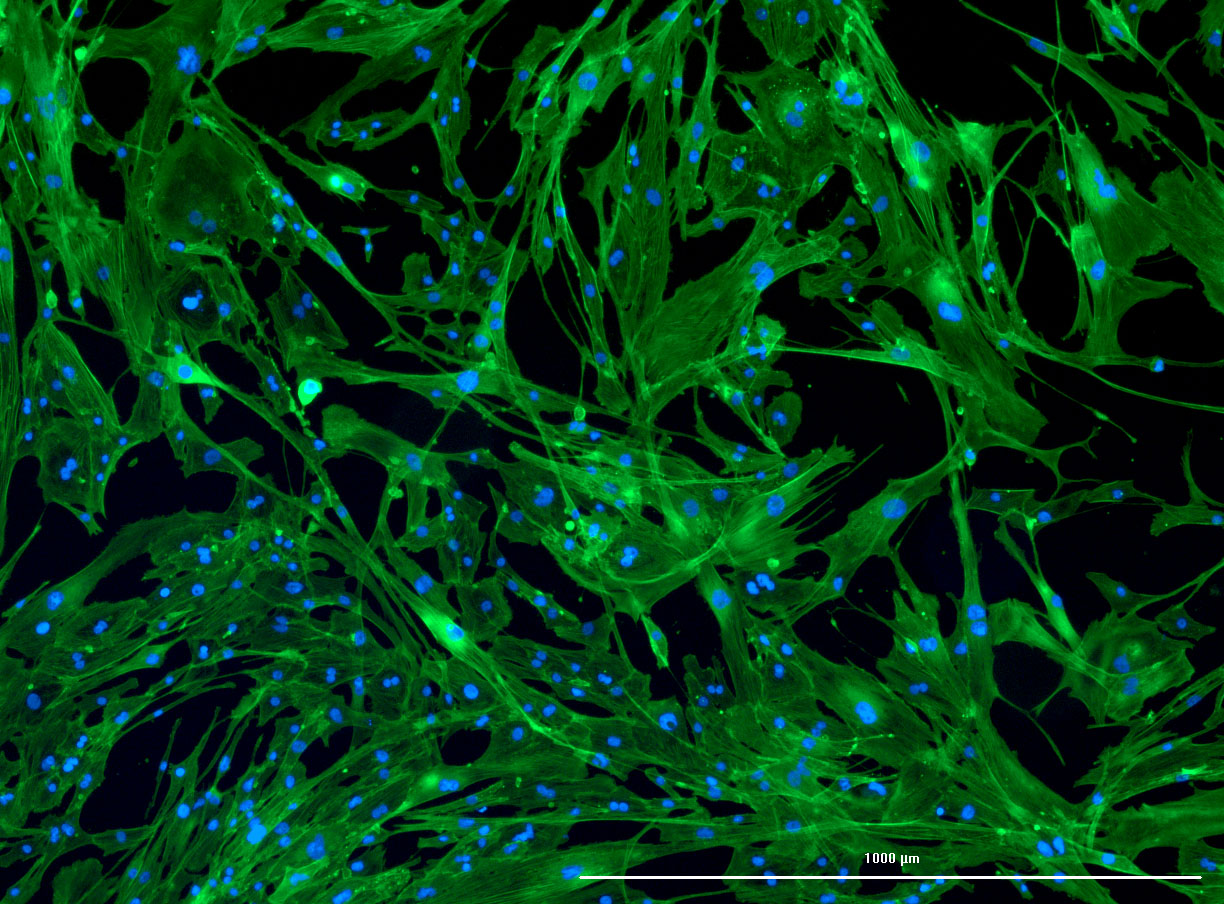
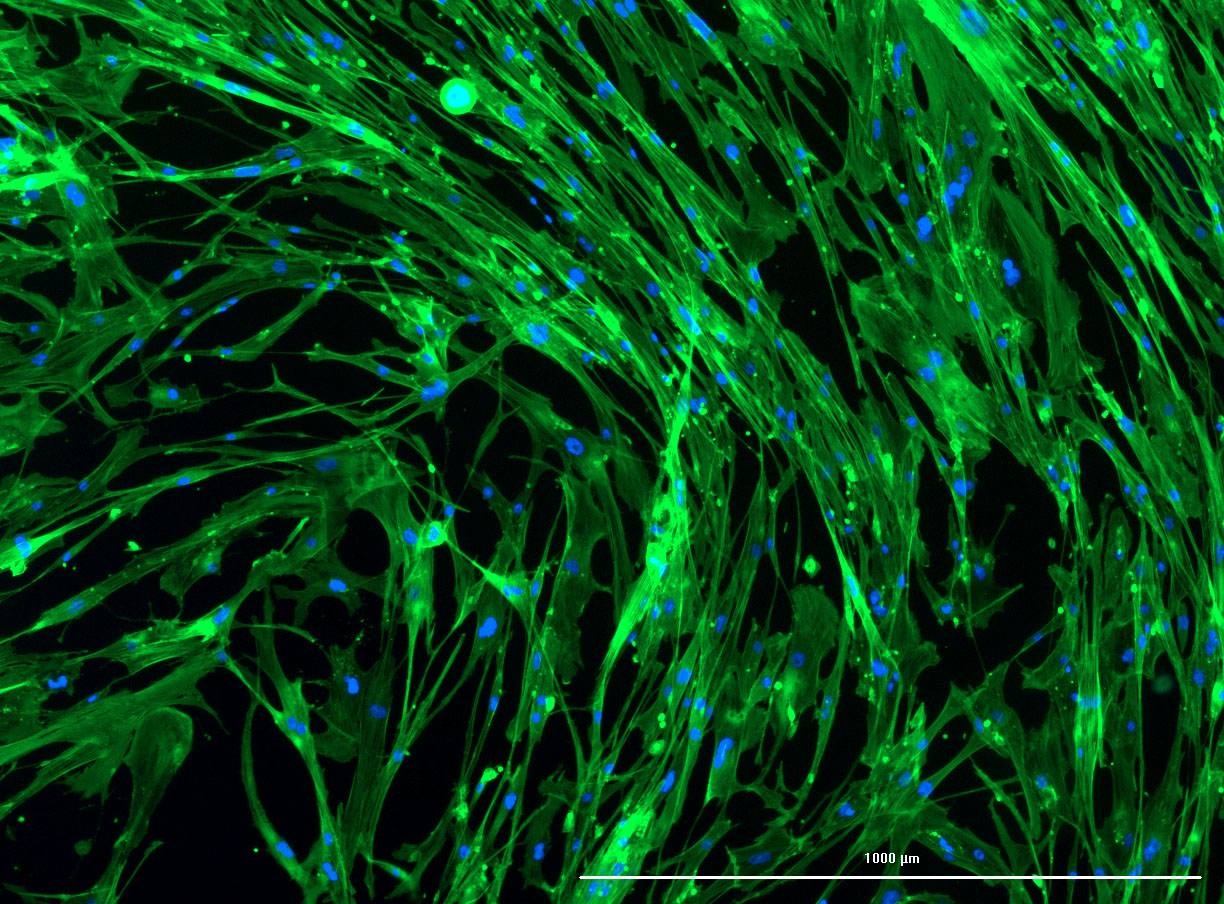
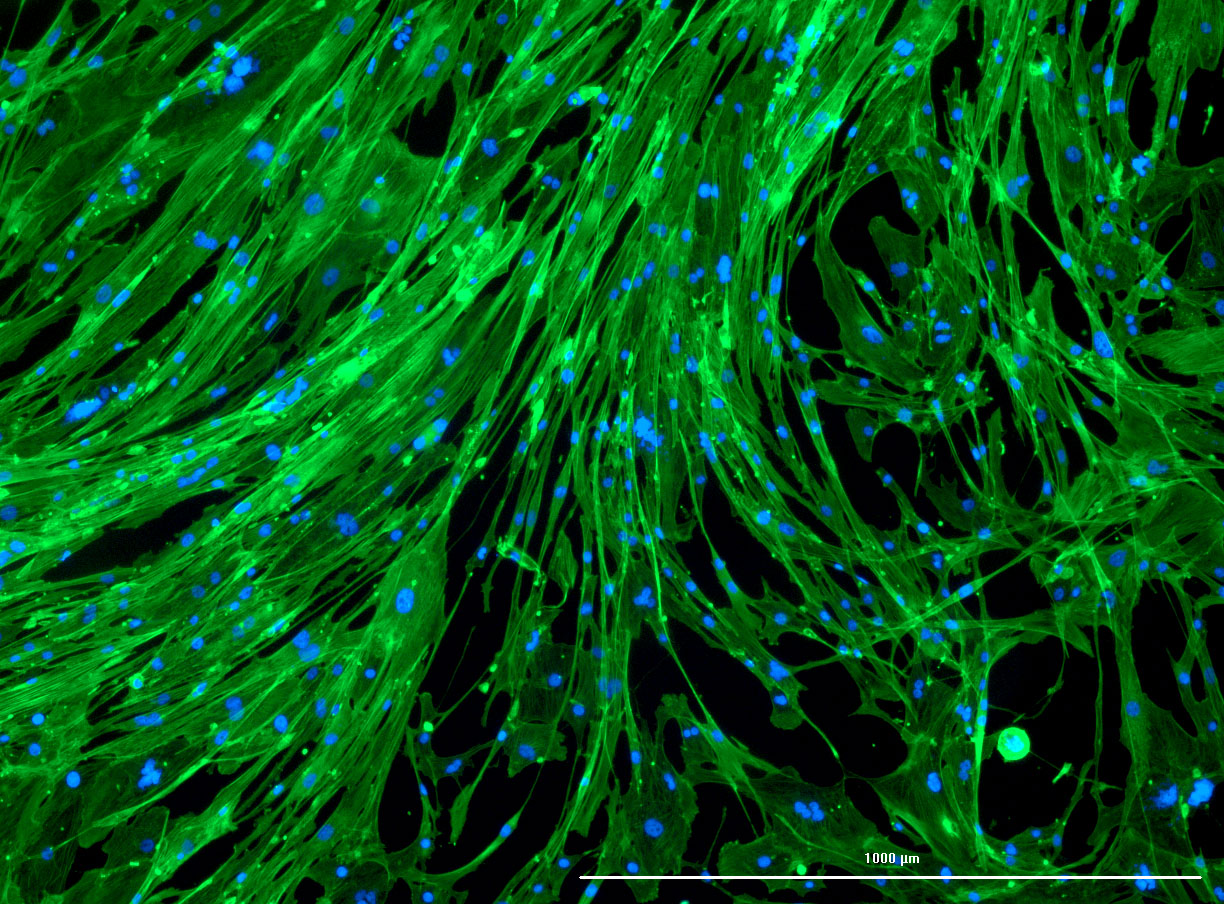
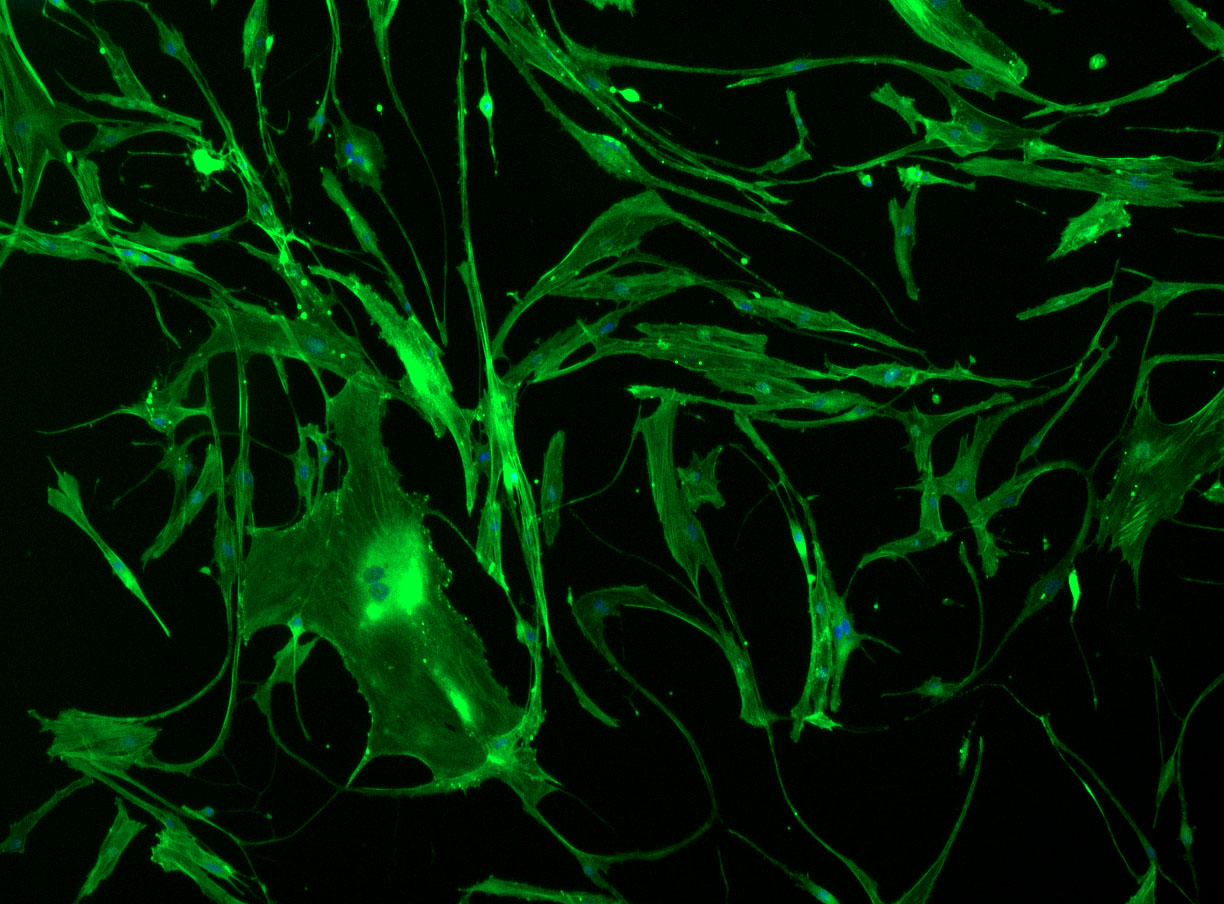
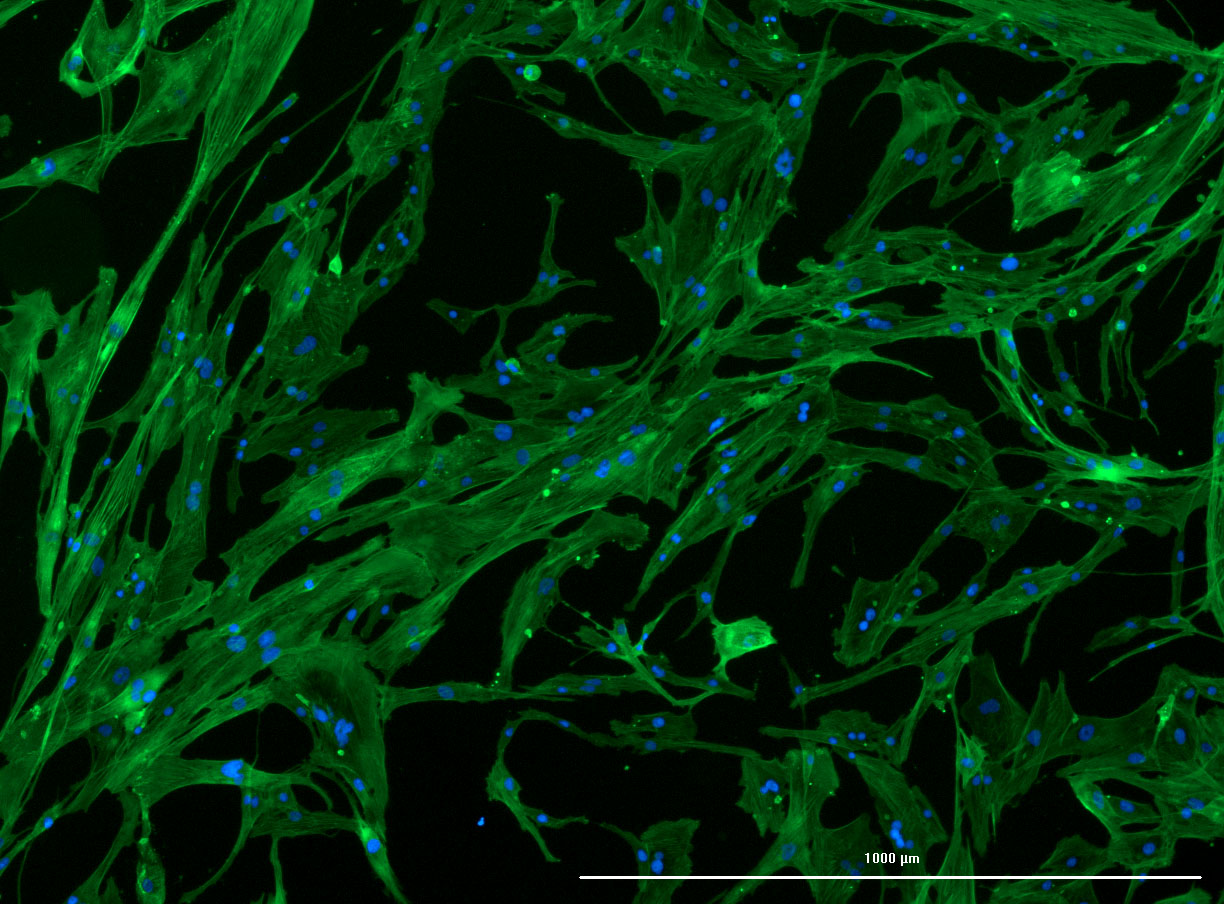 DAPI and Fluorescein Phalloidin staining of confluent fibroid cells P4 (although this is potentially misleading as the cells are very slow growing).
DAPI and Fluorescein Phalloidin staining of confluent fibroid cells P4 (although this is potentially misleading as the cells are very slow growing). 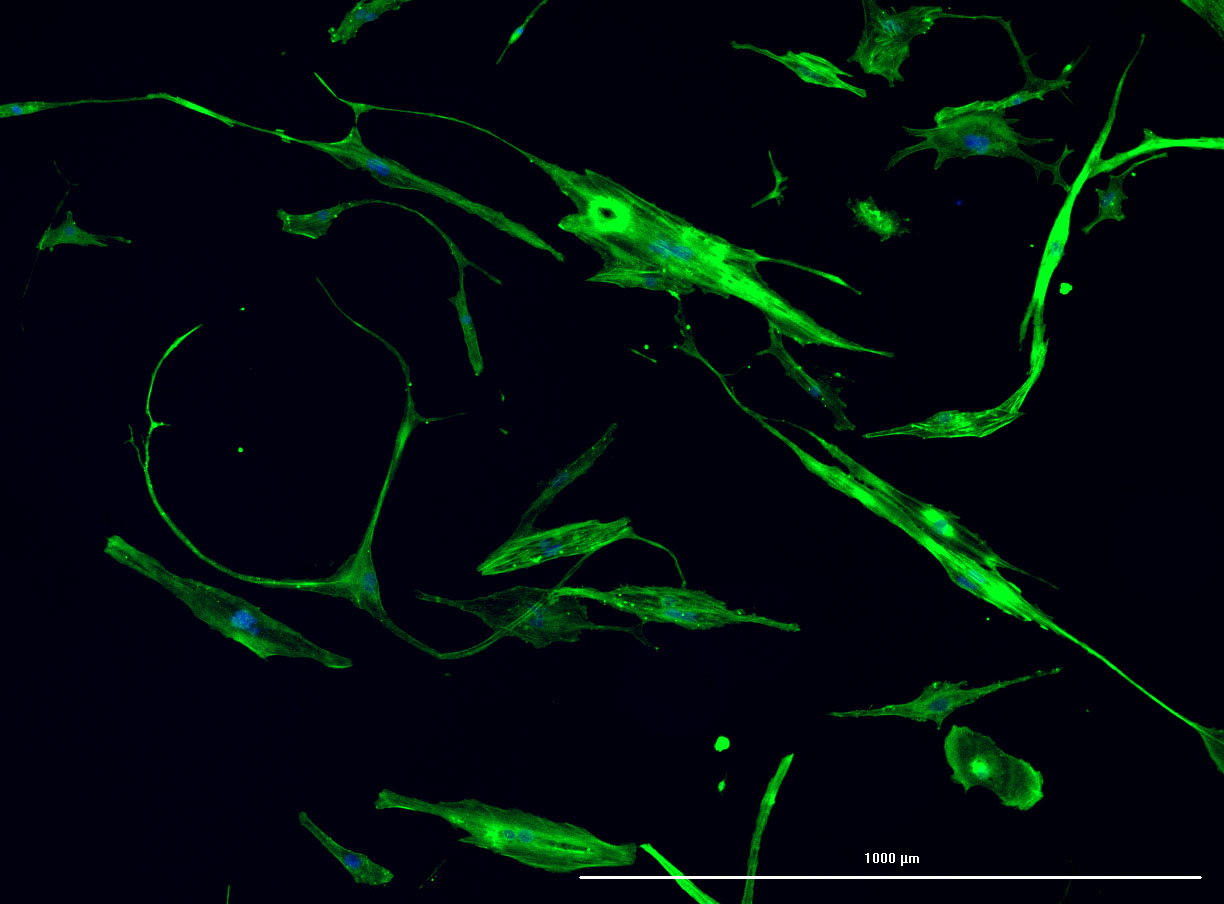
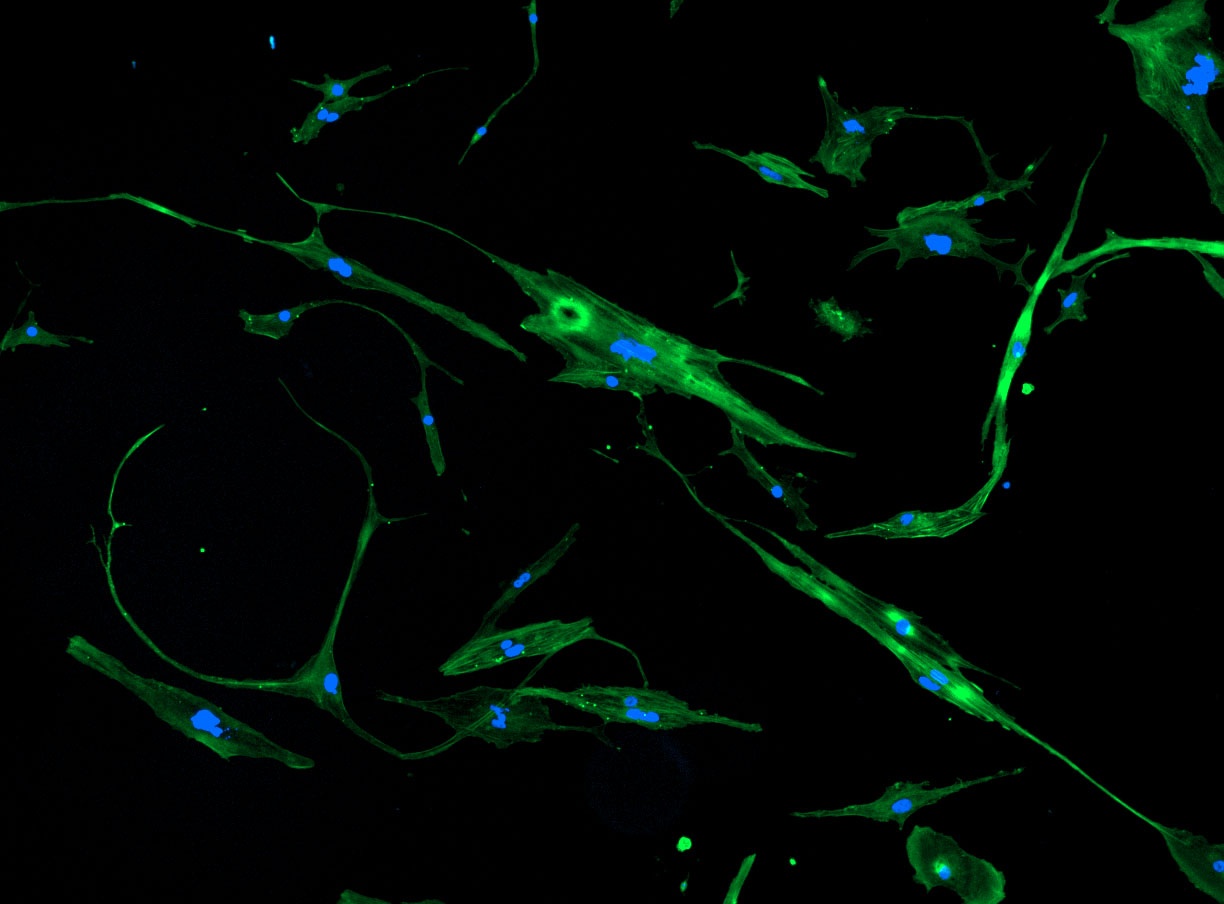
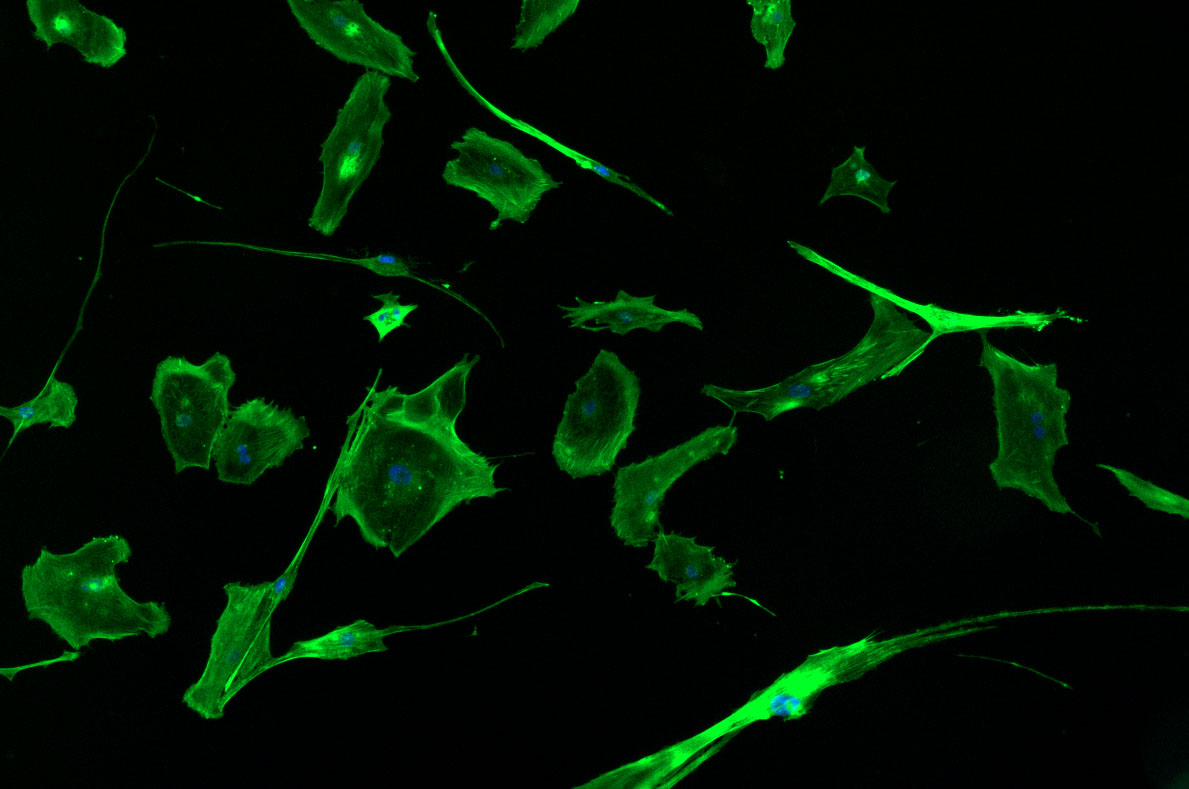
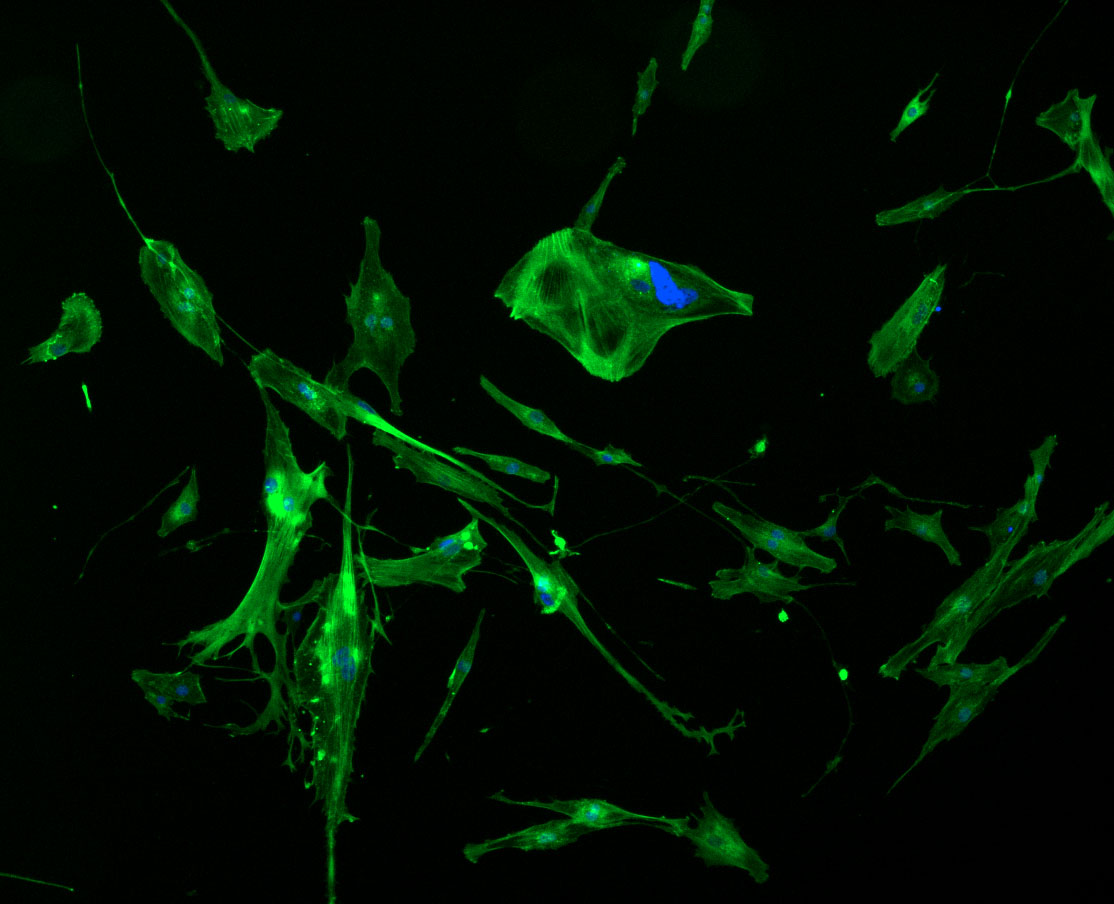
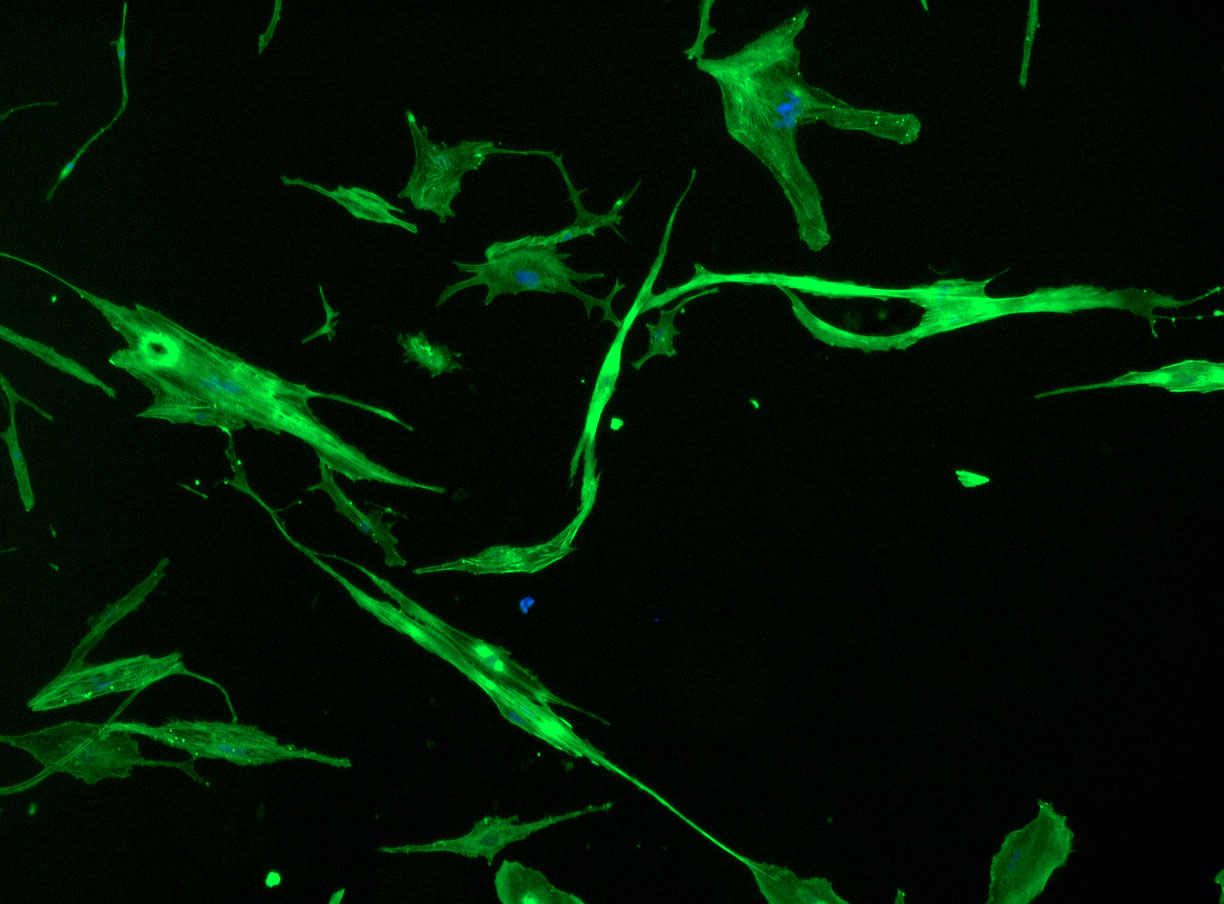 DAPI and Fluorescein Phalloidin staining of fibroid cells P4 which enables better visualisation of individual cells.
DAPI and Fluorescein Phalloidin staining of fibroid cells P4 which enables better visualisation of individual cells.  Working with 4% PFA in Fume Hood
Working with 4% PFA in Fume Hood Working at lab bench in the Stroke Group area
Working at lab bench in the Stroke Group area

 Unwrapping Petri dishes and getting ready to coat culture glassware with PLL.
Unwrapping Petri dishes and getting ready to coat culture glassware with PLL.  PLL coated glassware in Petri dishes ready for incubation.
PLL coated glassware in Petri dishes ready for incubation.  Cut glass vessels with cells ready for incubation.
Cut glass vessels with cells ready for incubation. 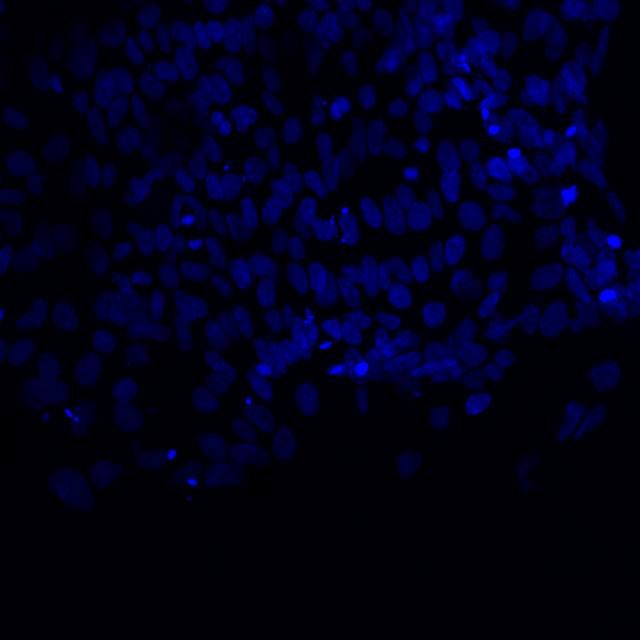 Microscopic image of stem cells, Hues 9 stained with DAPI (blue) by the UC San Diego Stem Cell Program.
Microscopic image of stem cells, Hues 9 stained with DAPI (blue) by the UC San Diego Stem Cell Program. Image of Amanita phalloides via
Image of Amanita phalloides via 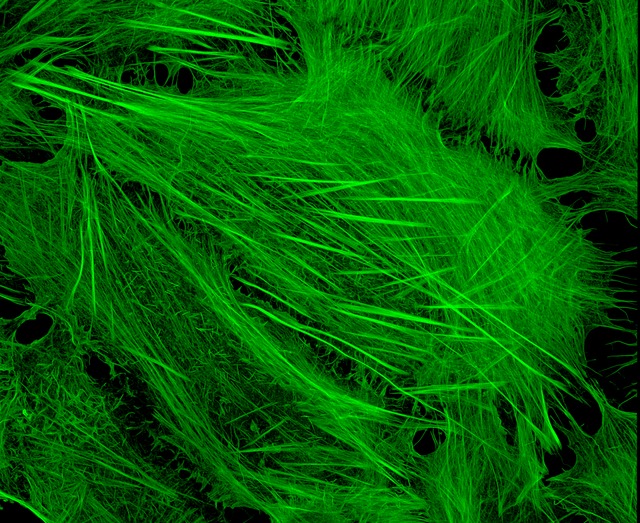 U2OS cells stained with fluorescent phalloidin taken on a confocal microscope by Howard Vindin
U2OS cells stained with fluorescent phalloidin taken on a confocal microscope by Howard Vindin Clear cut glass dish – approx 12cm diameter with central star motif and radiating pattern.
Clear cut glass dish – approx 12cm diameter with central star motif and radiating pattern. Cut glass dish and wrapped dish ready for sterilisation.
Cut glass dish and wrapped dish ready for sterilisation. Selection of glass vials for cell culture trial including metal closures.
Selection of glass vials for cell culture trial including metal closures.  Glass vessels in autoclave bags ready for sterilisation.
Glass vessels in autoclave bags ready for sterilisation.  Petri dishes and other glass items in autoclave bags ready for sterilisation.
Petri dishes and other glass items in autoclave bags ready for sterilisation. 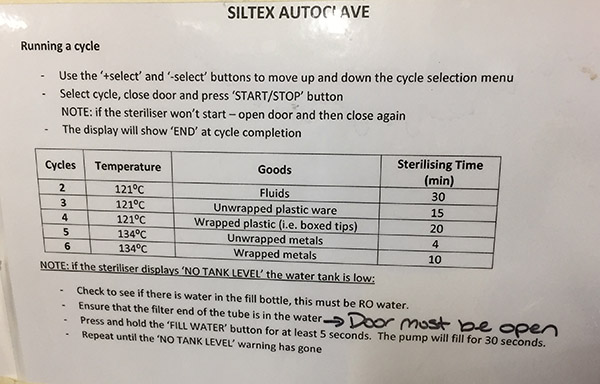 Autoclave instructions with cycle details.
Autoclave instructions with cycle details.  Autoclaved bags containing sterilised Petri dishes.
Autoclaved bags containing sterilised Petri dishes.  Autoclaved bags stored in lab area, ready for use.
Autoclaved bags stored in lab area, ready for use.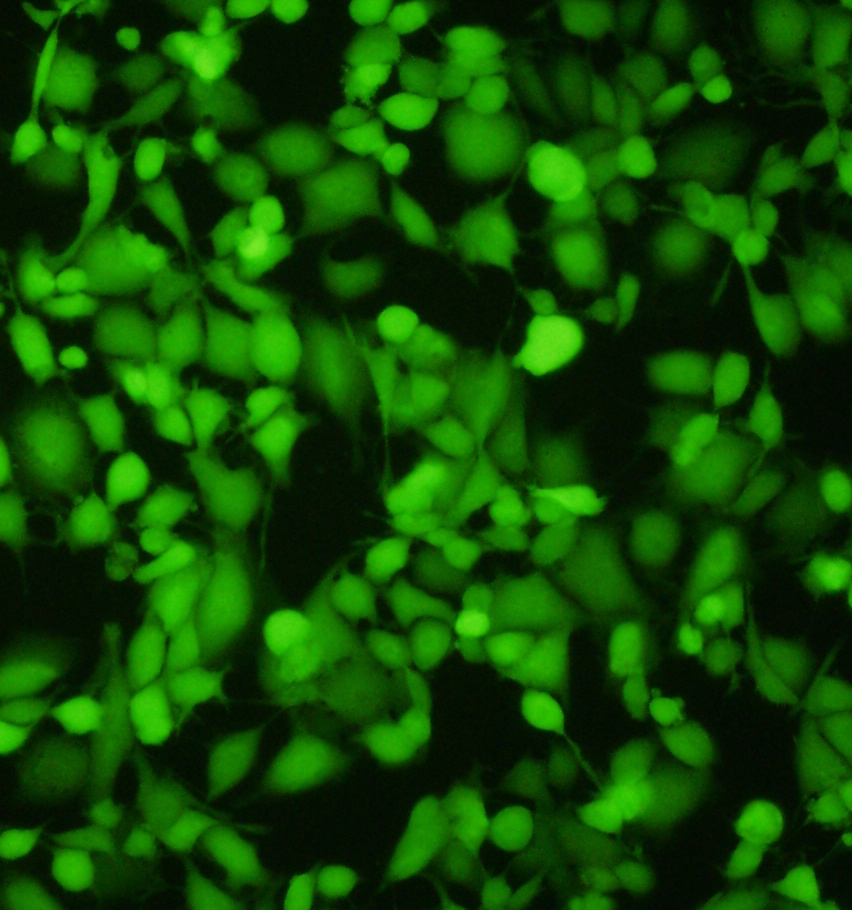 Impressive image of Calcein dye – live cells fluoresce a vibrant green – image via
Impressive image of Calcein dye – live cells fluoresce a vibrant green – image via 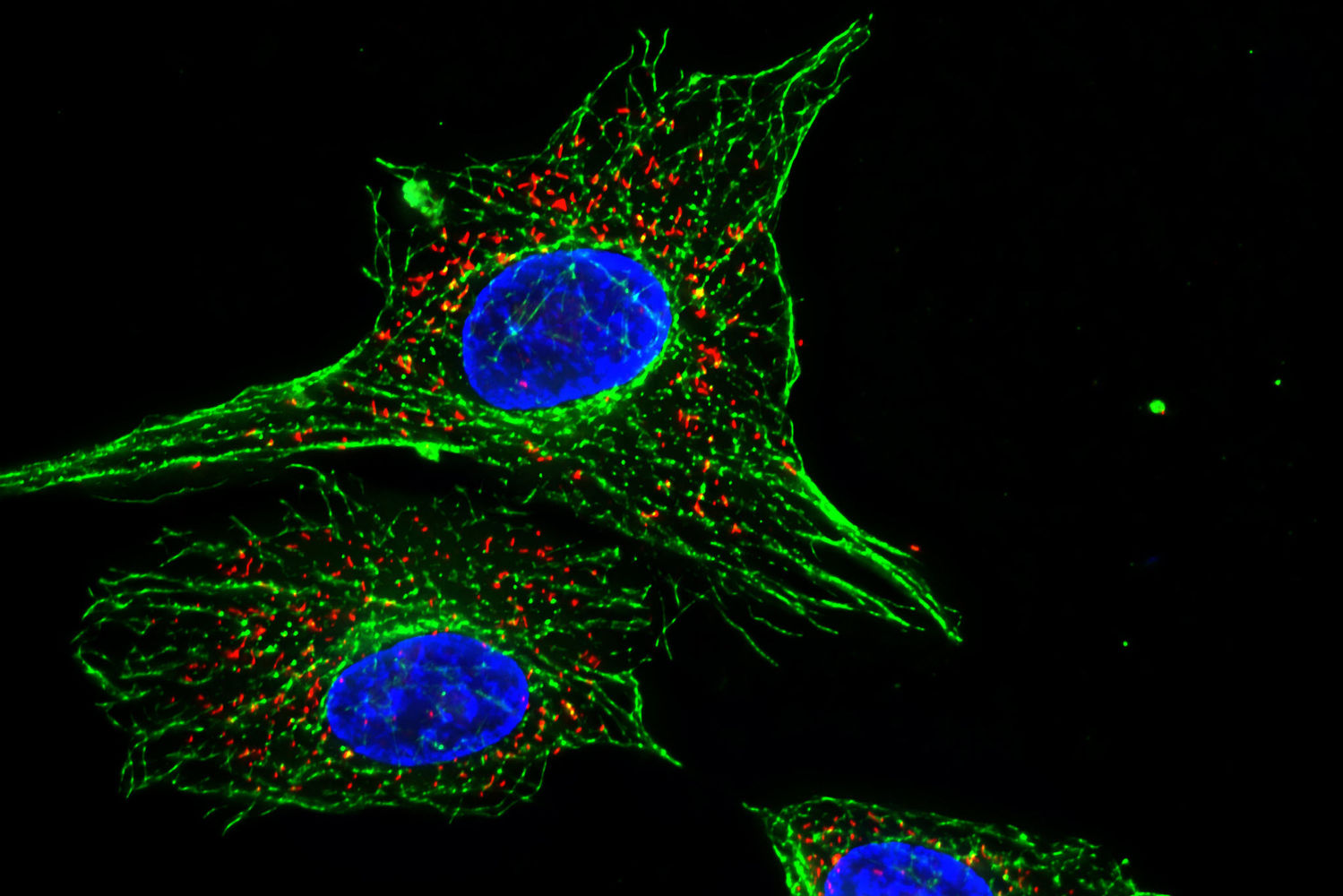 Image of fluorescent cells via
Image of fluorescent cells via {kind=link}