I’m gearing up for the holidays, with one final task completed – submission of HREC ethics amendment for the collection of blood for iPSC generation. Since the last amendment went though pretty quickly, I am hoping to have approval by the time I start back in the lab in early Jan – fingers crossed.
Monthly Archives: December 2021
Immunostained Cells
The lab has a great set-up for fluorescence microscopy which makes imaging quick and easy.
You just need to load the well plate into the machine and set up basic imaging parameters. You do need to image both DAPI and Phalloidin stains, but the software merges the images for you.
Fluorescent Images of Fibroid Cells
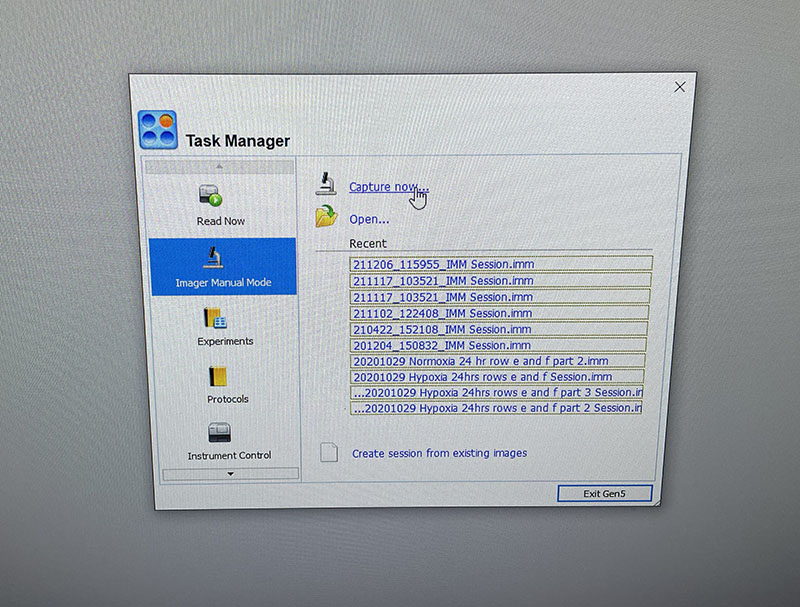
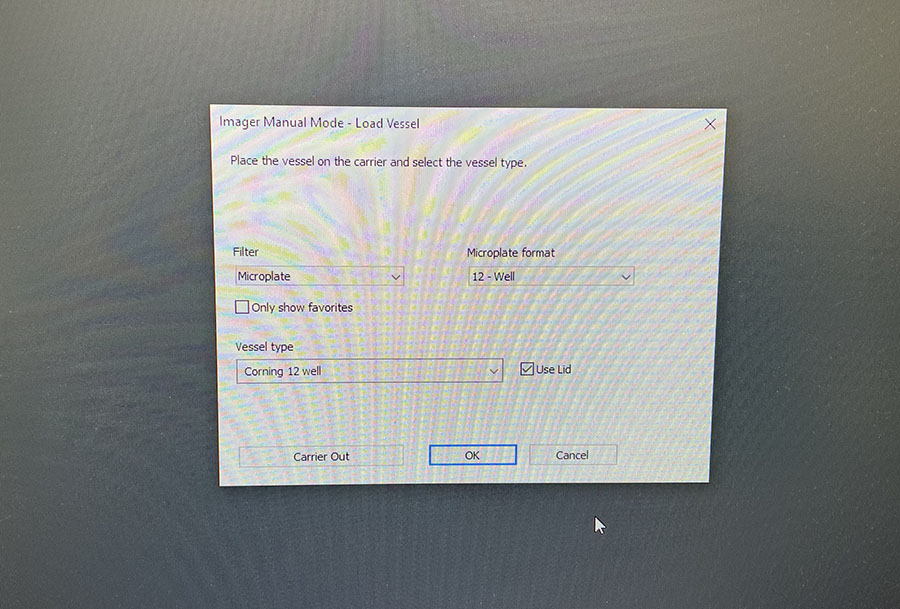 Simple graphic interface with presets ready to complete fluorescent microscopy.
Simple graphic interface with presets ready to complete fluorescent microscopy.
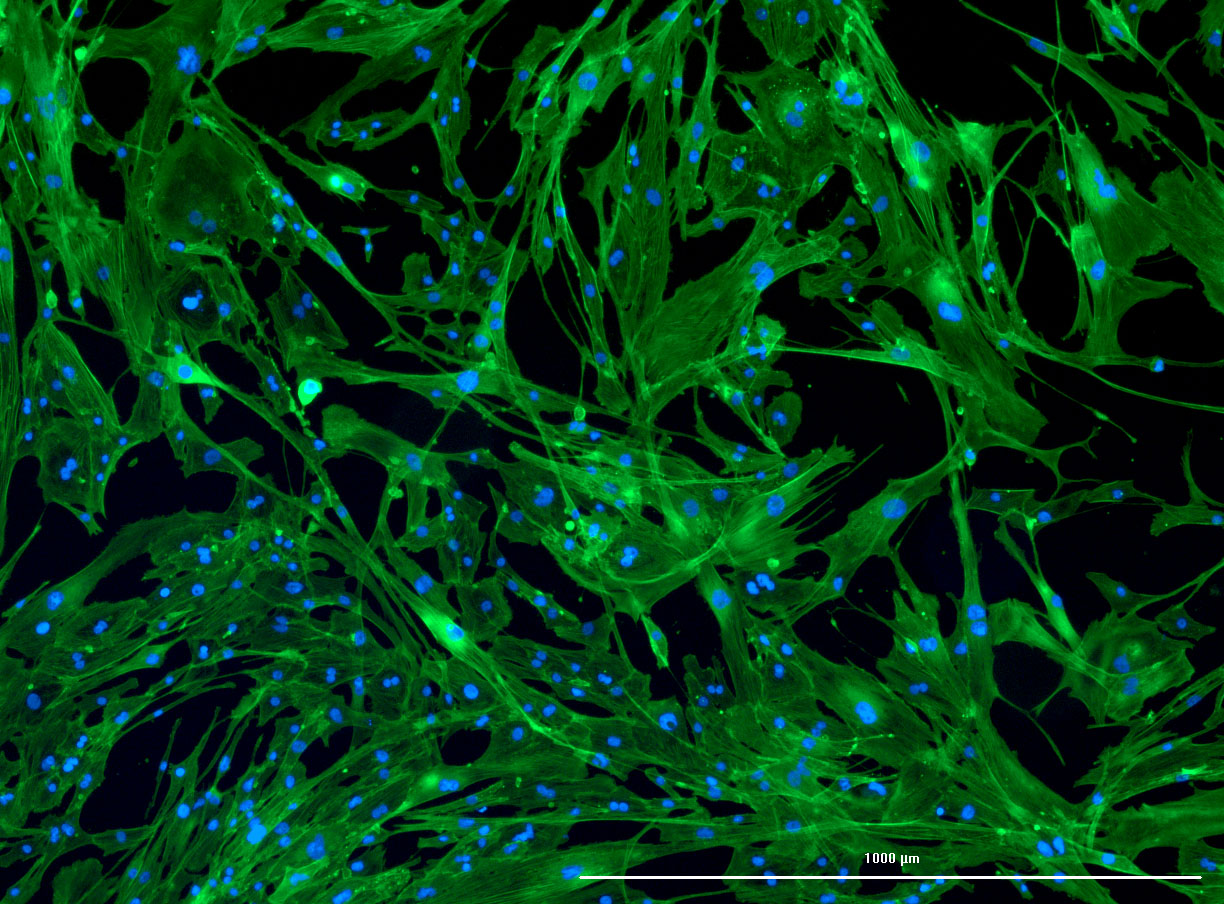
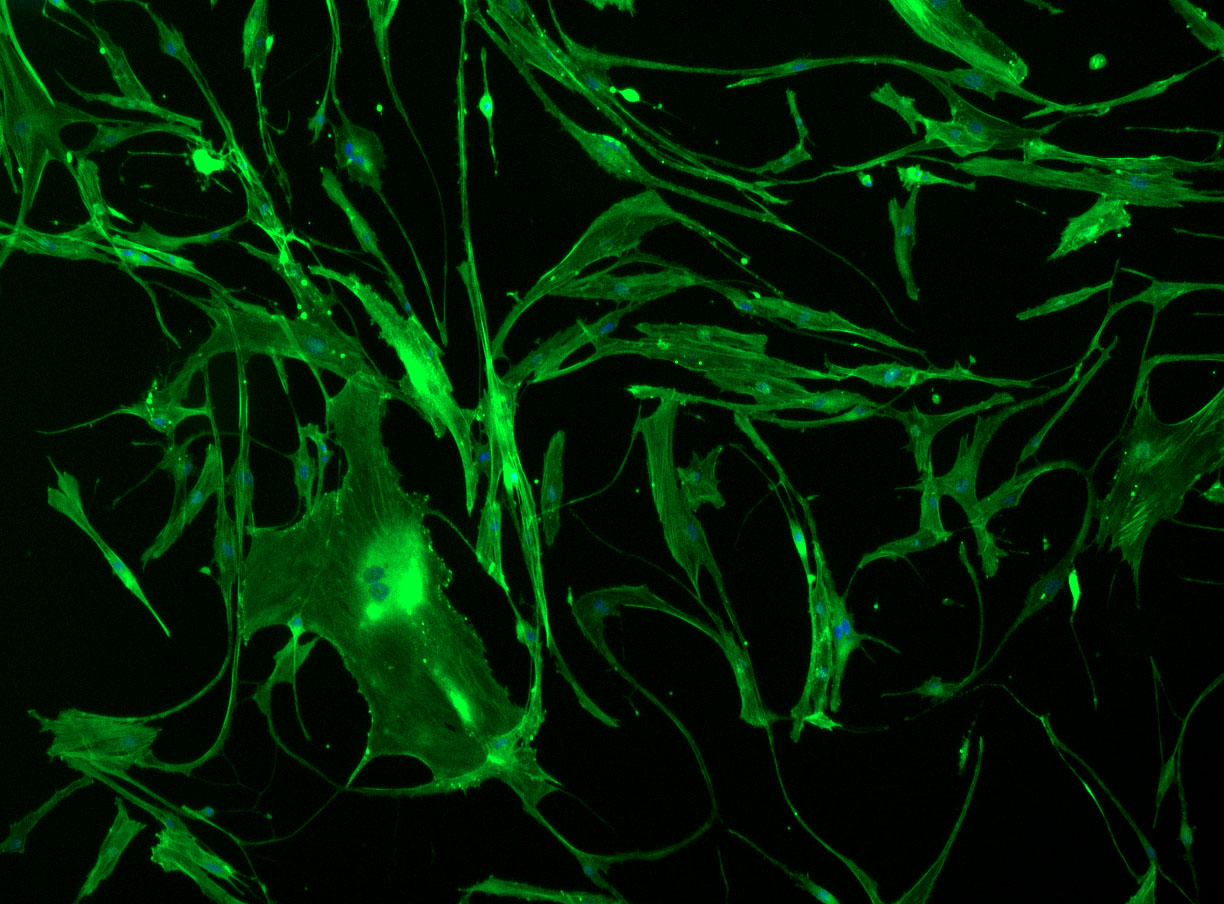
As discussed in my previous post on immunostaining, the blue dots indicate nuclei and the green structures reveal the cytoskeleton via binding to actin.
Confluent wells:
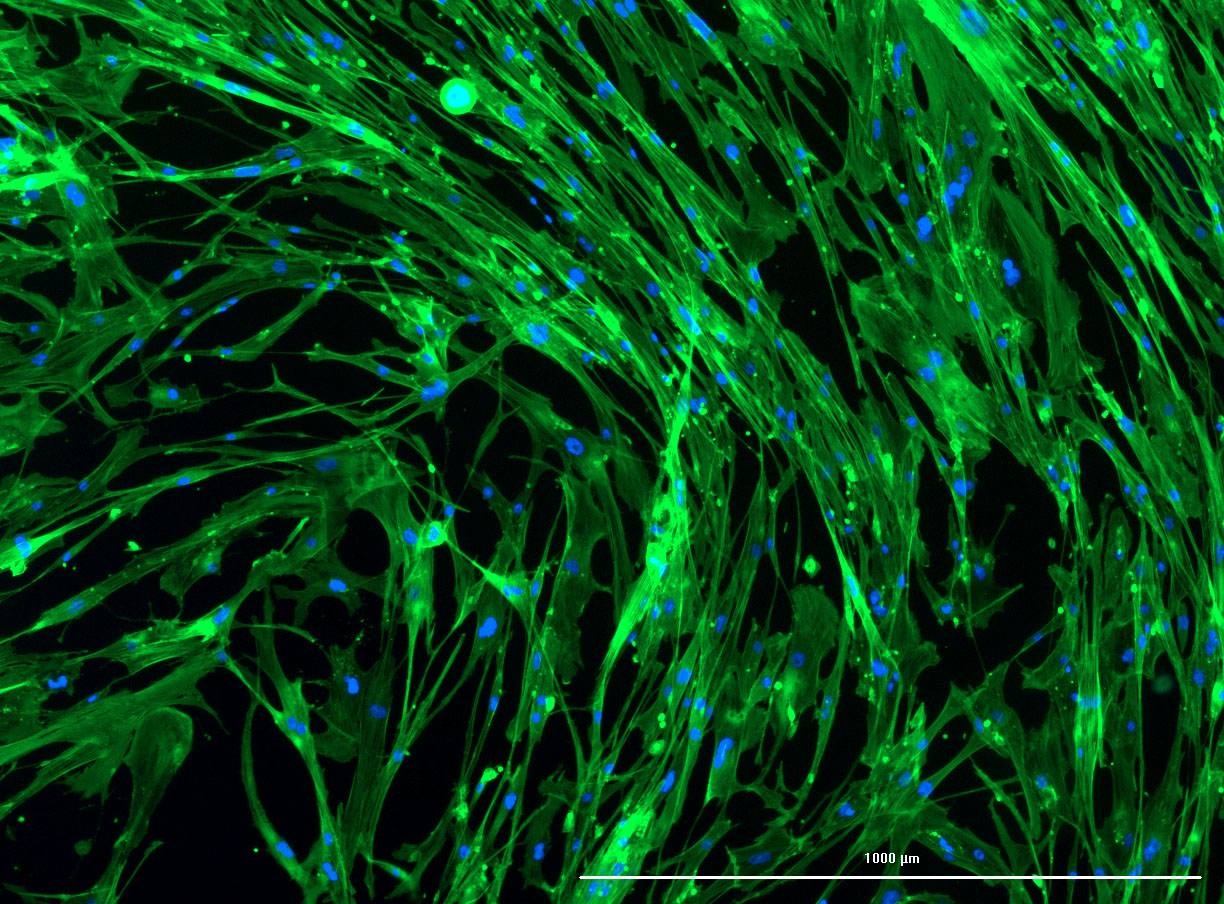
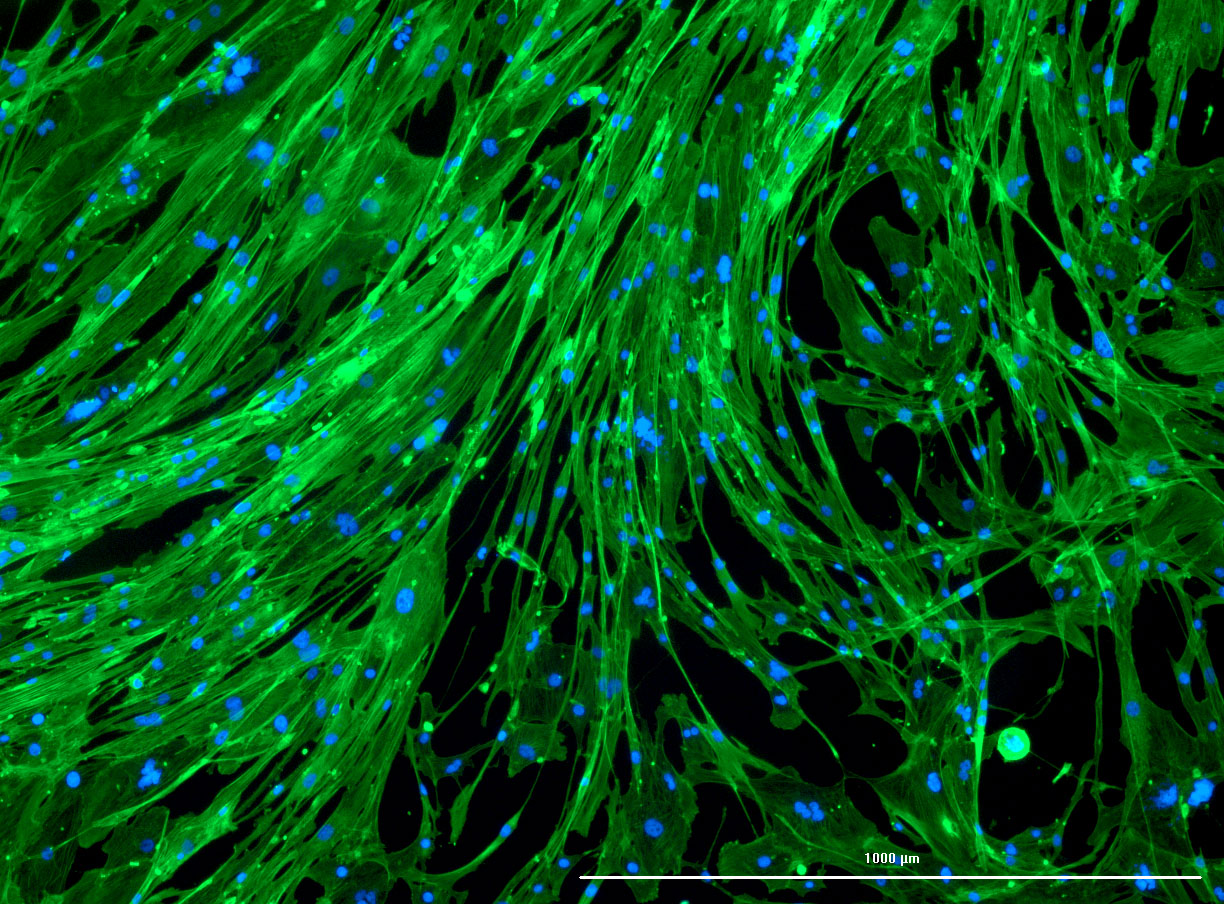
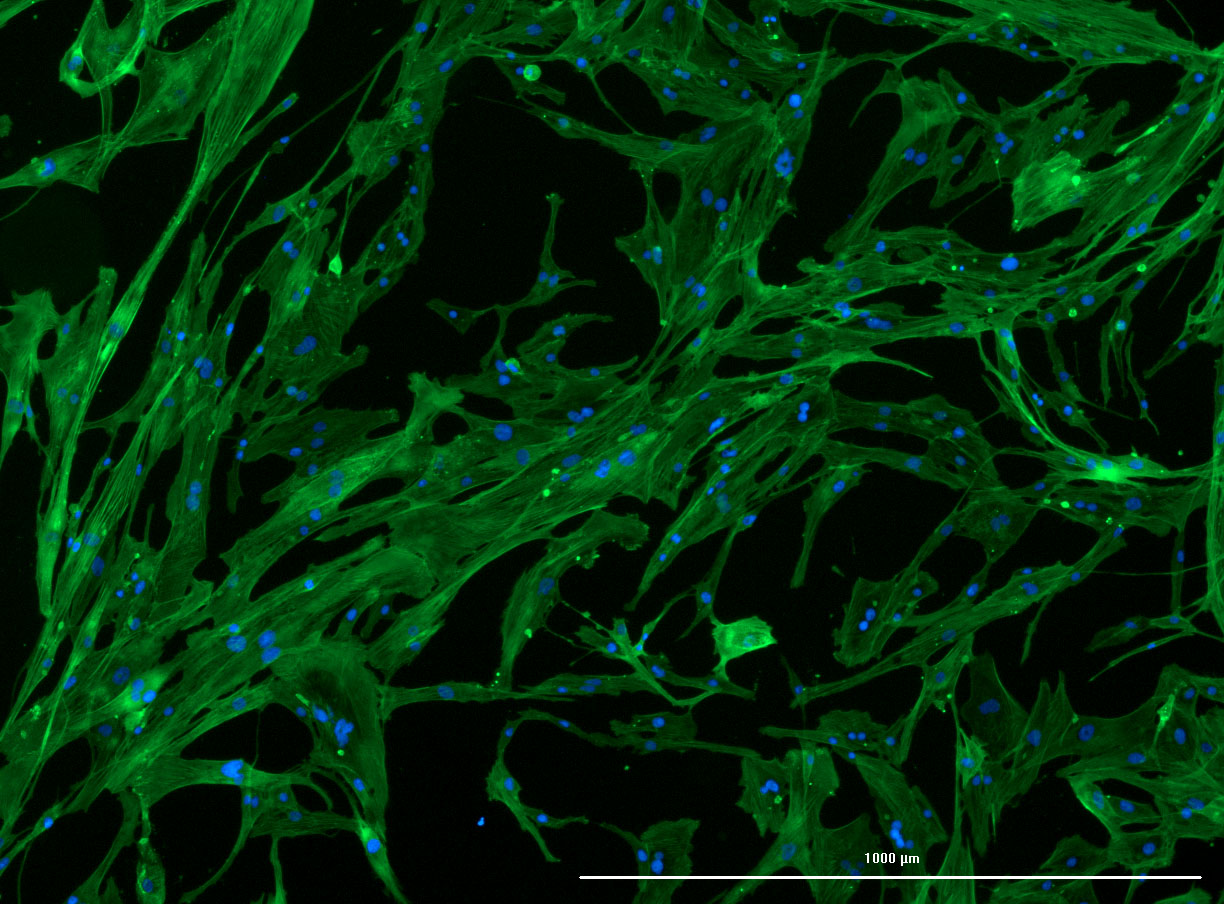 DAPI and Fluorescein Phalloidin staining of confluent fibroid cells P4 (although this is potentially misleading as the cells are very slow growing).
DAPI and Fluorescein Phalloidin staining of confluent fibroid cells P4 (although this is potentially misleading as the cells are very slow growing).
Less confluent wells:
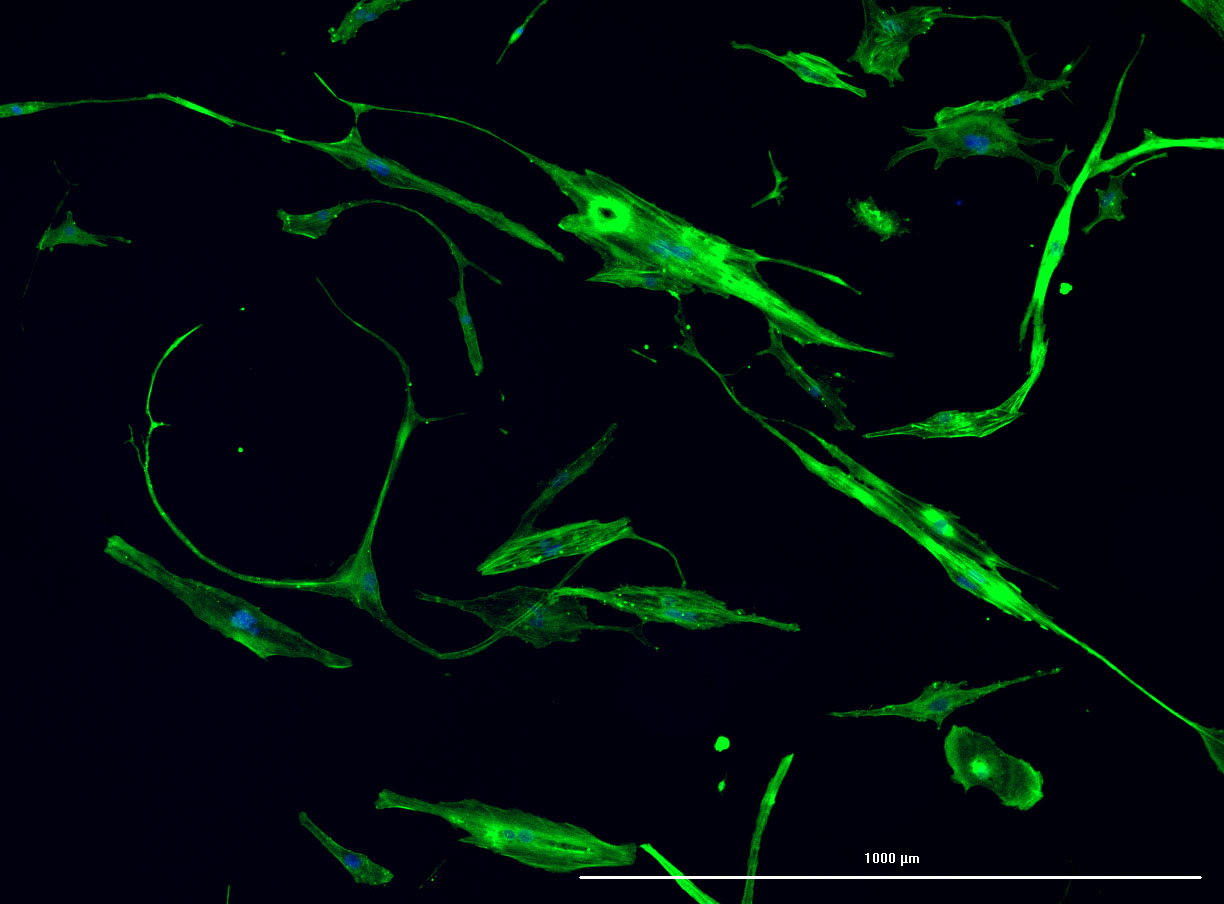
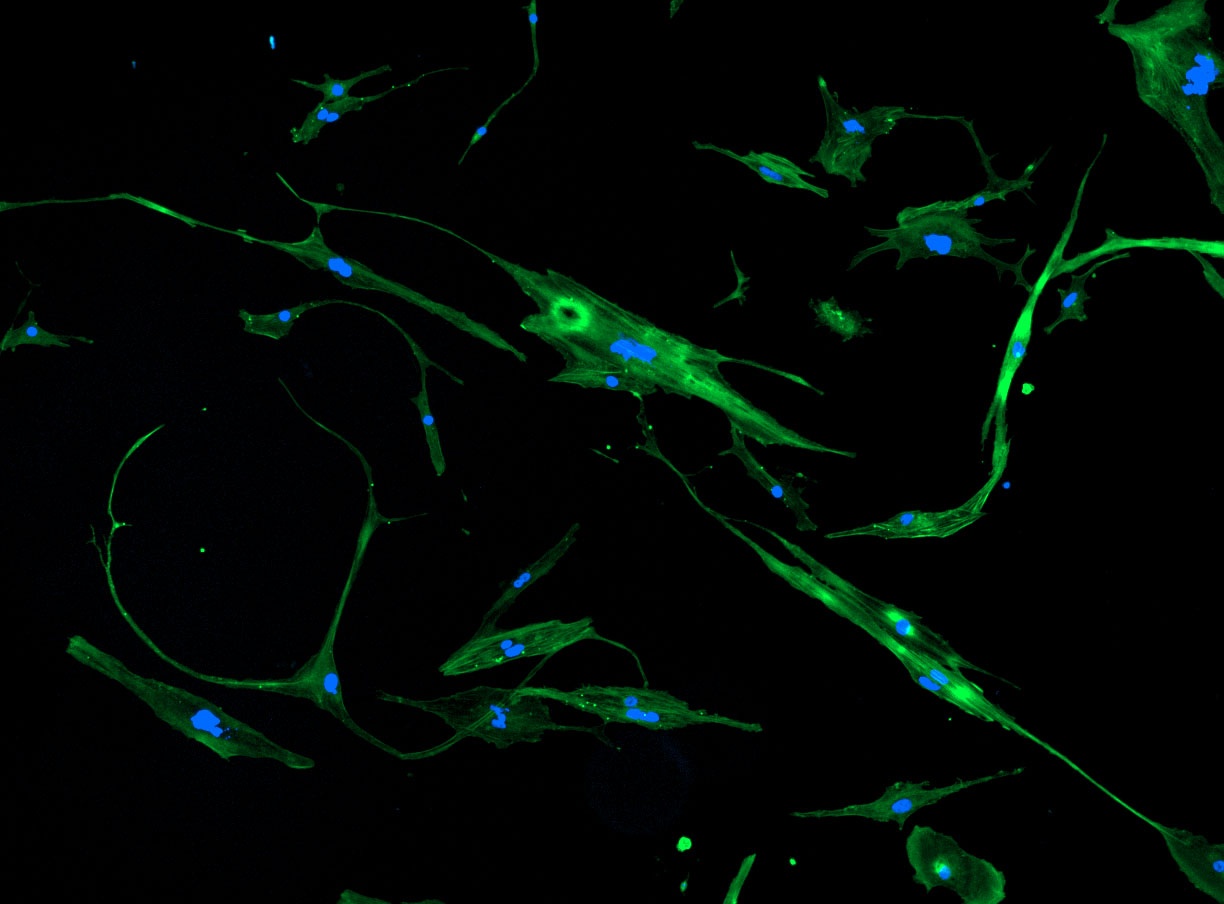
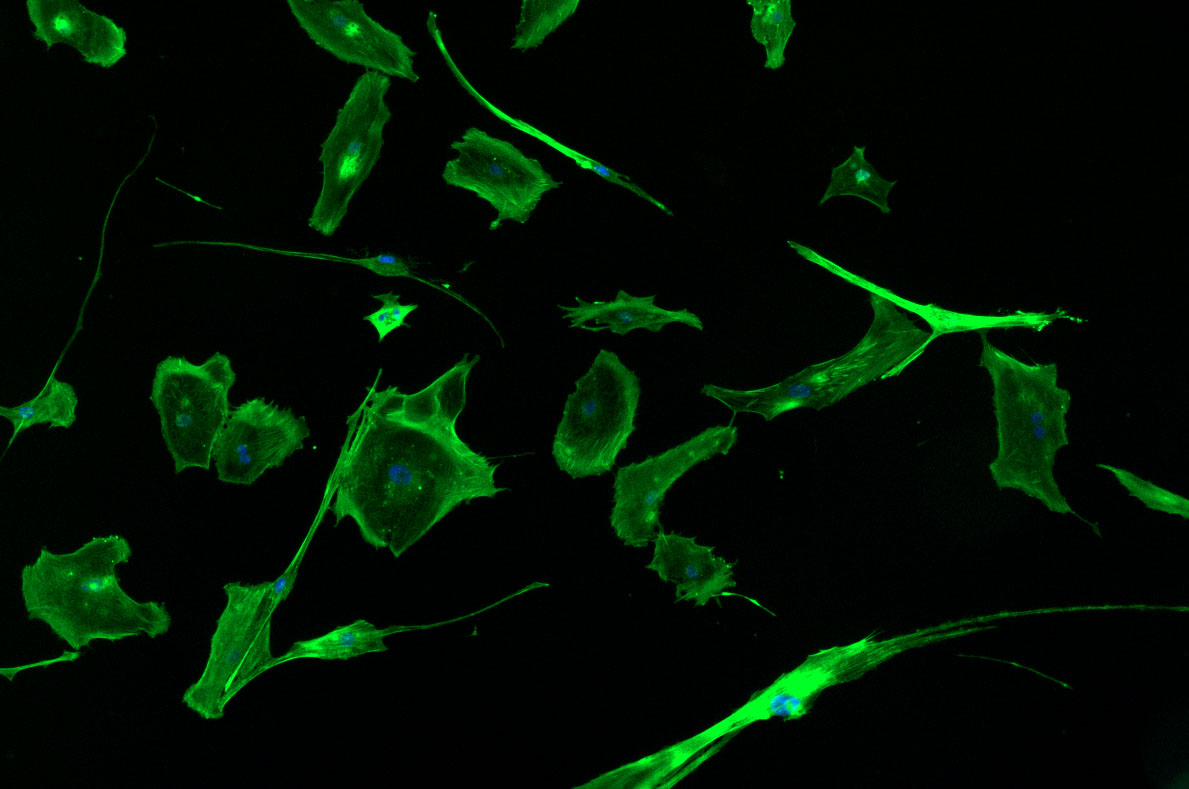
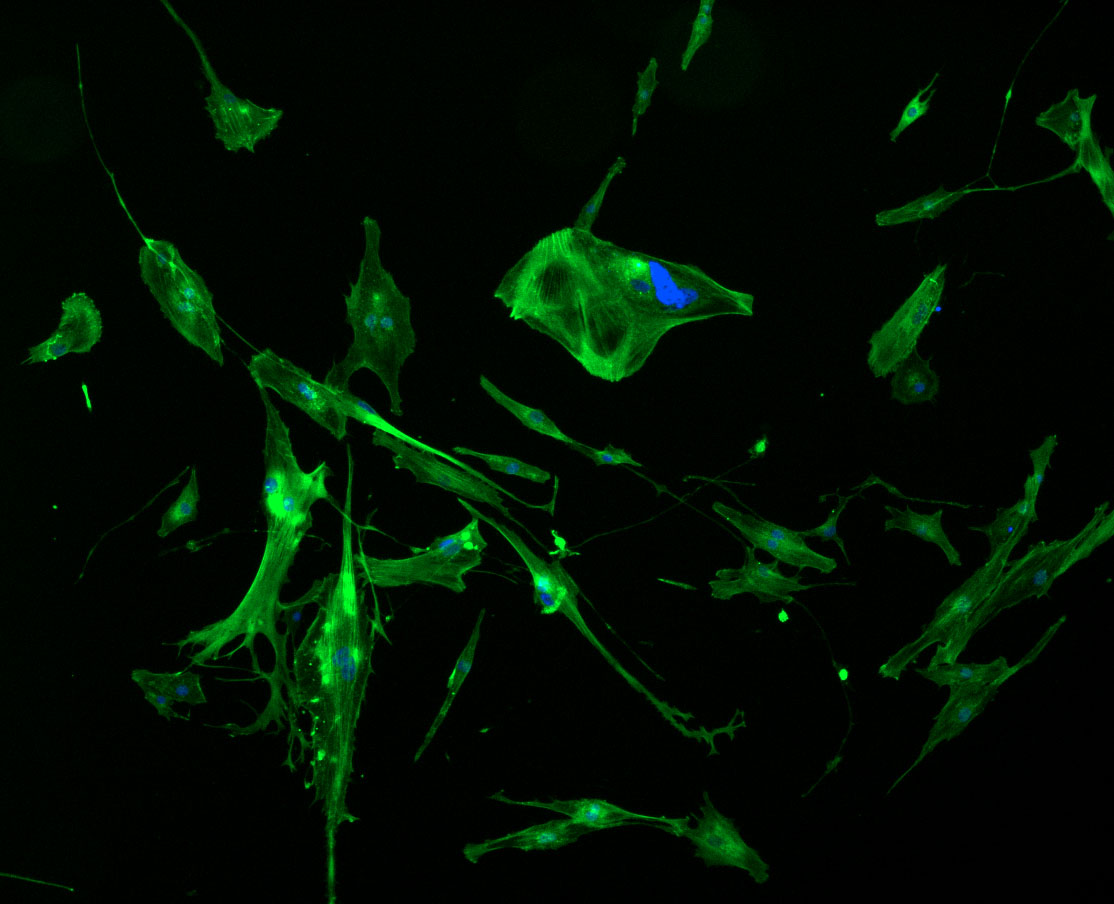
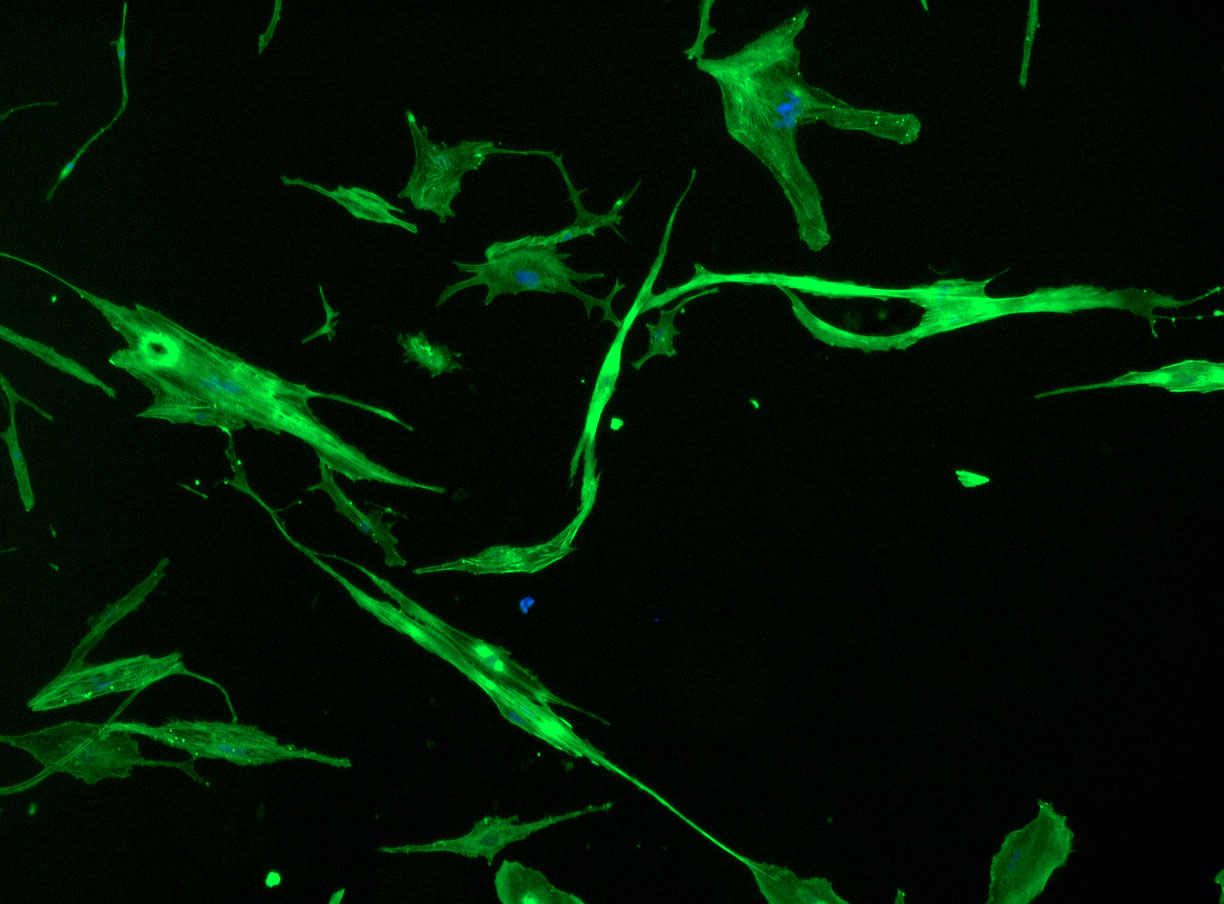 DAPI and Fluorescein Phalloidin staining of fibroid cells P4 which enables better visualisation of individual cells.
DAPI and Fluorescein Phalloidin staining of fibroid cells P4 which enables better visualisation of individual cells.
Immunostaining Protocol
It is time to complete the immunostaining protocol with guidance from Jo-Maree. I must admit that with holidays looming, my note taking was a bit sketchy. I will need to follow up with Jo-Maree to record the correct details of DAPI and Fluorescein Phalloidin stain. This will ensure that I know how to prepare (and order) stocks in the future.
I already prepared a couple of wells at different cell concentrations ready for staining. We had to delay the protocol, so some of the higher concentration wells are likely a bit over-confluent. It will be interesting to see how they look under the fluorescence microscope.
CELL FIXATION: ‘Dirty’ Biolab
 Working with 4% PFA in Fume Hood
Working with 4% PFA in Fume Hood
Prior to imaging, I fixed the cells in 4% PFA:
- Remove culture media (discard in waste container with bleach)
- Wash cells with PBS (discard in waste container with bleach) x 2
- Move cells to fume hood
- Add 4% PFA to each well for 15 – 20min at room temp (in fume hood)
- Remove 4% PFA solution (discard in PFA waste container in fume hood)
- Add PBS (make sure cells are covered or they dry out and produce poor images
IMMUNOSTAINING: At lab bench area
 Working at lab bench in the Stroke Group area
Working at lab bench in the Stroke Group area
- Remove PBS from each well
- Add 1mL 0.3% Triton X-100 (a strong detergent) to permeabilize cells (make cells permeable – this allows the phalloidin stain to enter the cell structure) for 10 min
- Make up DAPI (5mL PBS Tween and 1ul DAPI) and protect from light with aluminium cover
- Remove Triton X-100 and add 1mL DAPI solution to each well and incubate at room temp (with aluminum cover to protect from light) for 5 minutes

- Make up Flouroscein Phalloidin (1mL PBS sand 2μL Flouroscein) and protect from light
- Remove and discard DAPI solution
- Add 1mL PBS 0.1% Tween to each well for 5 min then discard x 3 (i.e. wash with PBS Tween x 3)
- Add Phalloidin stain to each well.
- Incubate at room temp for 1 – 2 hours
- Remove Phalloidin solution
- Wash with PBS Tween x 2
- Add 1mL PBS in each well
- Cover with aluminum foil to protect from light.
- Cells are ready for imaging. They can be stored in the fridge (with aluminium foil cover) until ready.
Growing my own cells in Petri Dishes
Following the successful growth of HBVPs in Poly-L-Lysine coated glass Petri dishes, I have enough of my own fibroid cells to repeat the process.
My cells continue to grow so slowly that I should be able to passage them into the Petri dishes and allow them to grow to confluence during the festive season break over 2 weeks . Of course, I need to clear this plan with Jo-Maree. No one else is using the incubator, so it should not be too much of a problem.
As part of this plan, I will be growing my tumour baby cells in 90mm glass Petri dishes and 1 x 90mm crystal dish. As per my previous experiment with HBVP cells, I need to coat the glass surface with Poly-L-Lysine solution to enable cell adherence.
I diluted the Poly-L-lysine solution with sterile MilliQ water (sterilised 14/12/21) to make up 40 mL total (10mL for each 900mm Petri dish x 3, plus 1 x cut glass crystal dish)
6mL PLL + 34mL MilliQ = 40mL PLL Solution

I added 10mL of the Poly-L-Lysine solution to each dish and then incubated them for an hour. [ The cut glass crystal dish was placed inside a 150mm autoclaved Petri dish to preserve sterility.]
 Unwrapping Petri dishes and getting ready to coat culture glassware with PLL.
Unwrapping Petri dishes and getting ready to coat culture glassware with PLL.
 PLL coated glassware in Petri dishes ready for incubation.
PLL coated glassware in Petri dishes ready for incubation.
Following incubation, I removed the Poly-L-Lysine solution and washed the dish with PBS. During cell passage of my confluent flask, I added 1mL of cell solution (from a 10mL suspension) and 5mL media. I placed the cut glass vessels back into a 150 mm Petri dish and into the incubator.
 Cut glass vessels with cells ready for incubation.
Cut glass vessels with cells ready for incubation.
Fingers crossed that they survive the holiday break!
Immunostaining of fibroid cells
To gain some more insight into the cellular structure of my fibroid cells, I asked Jo-Maree to help me with immunostaining. Immunostaining refers to a staining method that uses antibodies to stain different proteins and structures within the cell. We are going to start with an easy protocol using two antibodies: DAPI and Phalloidin.
DAPI (4′,6-diamidino-2-phenylindole) is a very common fluorescent blue stain used to reveal the nucleus in cultured cells. It can penetrate the intact membrane of the cell. Thus, it can be used for staining both fixed and live cells.
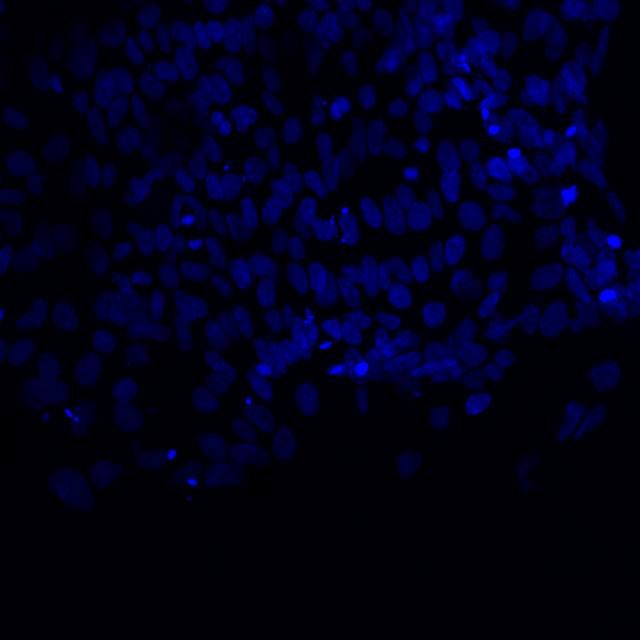 Microscopic image of stem cells, Hues 9 stained with DAPI (blue) by the UC San Diego Stem Cell Program.
Microscopic image of stem cells, Hues 9 stained with DAPI (blue) by the UC San Diego Stem Cell Program.
BASIC LIVE CELL STAINING PROTOCOL
- Add DAPI to the complete culture medium used for cultured cells at a concentration of 10 ug/mL.
- Remove culture medium from the cells and replace with the medium containing the DAPI.
- Cover cells from light exposure and incubate at room temperature or 37°C for 5-15 minutes, then image.
Direct Addition: According to the biotium protocol, you can also stain cells by adding the dye directly to the cell culture and medium. However, this requires a 10X concentration of dye.
- Add the dye to complete culture medium at 10 times the final recommended staining DAPI concentration – 100 ug/mL..
- Without removing the medium from the cells, add 1/10 volume of 10X dye directly to the well.
- Immediate mix thoroughly by gently pipetting the medium up and down. For larger well sizes (e.g., 24-well to 6-well plates), the plate can be gently swirled to mix.
- Cover cells from light exposure and incubate cells at room temperature or 37°C for 5-15 minutes, then image
STAINING FIXED CELLS/TISSUES:
- Add DAPI to PBS at 1 ug/mL.
- Add the PBS with dye to cells or tissue sections and incubate at room temperature for at least 5 minutes with covering from light exposure.
- Samples can be stored in a lightsafe covering (e.g. aluminium foil) at 4°C after staining and before imaging
From: https://biotium.com/tech-tips/protocol-staining-cells-with-hoechst-or-dapi-nuclear-stains/ with minor edits for simpliticy.
Since we are using phalloidin and fixing the cells prior to imaging, we need to follow some additional step to make the cells permeable.
Phalloidin Jo-Maree did not have any relevant stocks, Natalie kindly sourced a sample from another group. [I am always impressed by the generosity of functioning labs and how groups are happy to share stocks to enable other researchers to move forward.]
We are using Fluorescein Phalloidin as a counterstain to enable the visualisation of actin – a protein found in large quantities in the cytoskeleton and cell muscle fibres. As such, it plays a vital role in cell muscle contraction and overall cell movement.
Interestingly Phalloidin is a toxin (specifically phallotoxin) derived from Amanita phalloides (death cap mushroom).
 Image of Amanita phalloides via Wikimedia Commons.
Image of Amanita phalloides via Wikimedia Commons.
{kind=link}
Phalloidin binds very well to actin filaments and is therefore very useful in visualising cell structure.
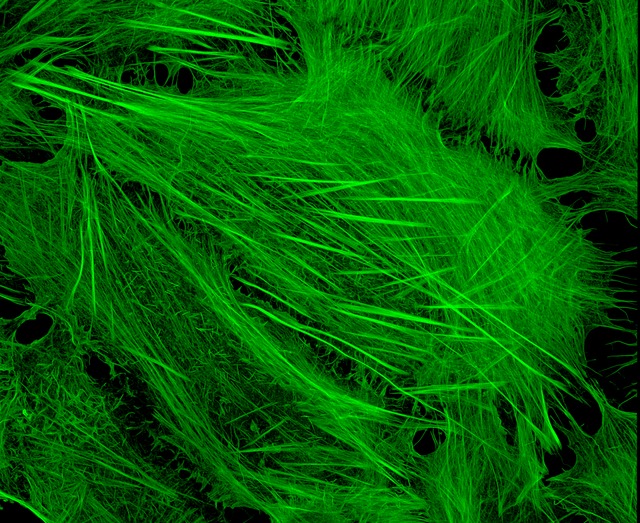 U2OS cells stained with fluorescent phalloidin taken on a confocal microscope by Howard Vindin
U2OS cells stained with fluorescent phalloidin taken on a confocal microscope by Howard Vindin
STAINING FIXED CELLS
- Fix cells in 3–4% formaldehyde in PBS at room temperature for 10–30 minutes.
- Remove fixation solution and wash cells 2–3 times in PBS.
- Add 0.1% Triton X-100 in PBS into the fixed cells for 3–5 minutes to increase permeability. Then wash cells 2–3 times in PBS.
- Add phalloidin-conjugate working solution. Incubate at room temperature for 20–90 minutes.
- Add DAPI DNA staining dye at this point.
- Rinse cells 2–3 times with PBS, 5 min per wash.
- Cover with lightfast material to preserve fluorescence
Adapted from: https://www.abcam.com/protocols/phalloidin-staining-protocol
Revised Ethics – Blood cells for iPSCs
Due to COVID supply issues, we are still having issues sourcing key reagents etc. for the project. As an alternative iPSC protocol, we are now planning on using blood cells. The main reason is that it is a regular and active protocol in the broader lab area with clear in-house expertise. This also works better conceptually for me than harvesting cells from a skin biopsy – after all blood is strongly associated with notions of kinship . It is also nice to move into the footsteps of my dear colleague Dr Trish Adams who used iPSC technology to turn blood cells into heart cells for the project Machina Carnis.
This does entail a further ethics amendment, but since we have prior approval for skin biopsy harvest, I do not foresee any major issues. I hope to submit this before holidays – ready for the new year!
AAANZ – Upcoming Panel

I am delighted to be part of a panel for the AAANZ conference on Friday 10 December 2021: Impact of exchange: axis of collaboration between the arts and sciences
This is a wonderful opportunity to share work and approaches with some amazing colleagues including Helen Pynor who is the current ANAT SAHMRI Resident working with Jimmy Breen.
I will provide an overview of my ongoing collaborative engagement with Prof. Dietmar Hutmacher who leads the Centre for Regenerative Medicine at QUT including an update on the ANAT residency project.
Abstract: What is a partnership between an artist and a scientist? What new knowledge do they impart on each other’s discipline, and does it change the way they work and think? This panel will bring to light the kinds of relationships that artists and scientist have, generating new knowledge, ideas and processes resulting in transdisciplinary outcomes that have continuing impacts in both the sciences and art. What can we learn from these relationships and how do we recognise that art/science interdisciplinary and collaborative practice has equal value in both fields? This panel will bring together researchers to discuss how these relationships form, and the kinds of questions artists and scientists bring to the table in order to collaborate. Therefore, we will address the challenges and problems of these experiences, but also how these partnerships redefine the parameters of interdisciplinary engagement and the future of such intersections.
Session Convenor
Dr Erica Seccombe, Australian National University
Gregory Minissale, University of Auckland
Session Speakers
Dr Helen Pynor, ANAT resident, SAHMRI Bioinformatics Platform
Dr Jimmy Breen, Robinson Research Institute, University of Adelaide
Dr Svenja J. Kratz, University of Tasmania
Dr Baden Pailthorpe, Australian National University
Dr Tony Curran, Independent Artist and Scholar
Gregory Minissale, University of Auckland
Cut Glass Collection
As part of the residency project, I have started a collection of cut glass items. These were sourced from different second hand shops and build on an existing collection of items used for an exhibition at The Edge at the State Library of Queensland in 2013.
I am particularly attracted to the patterns of the glass. A recurring central motif in many items is a star.
 Clear cut glass dish – approx 12cm diameter with central star motif and radiating pattern.
Clear cut glass dish – approx 12cm diameter with central star motif and radiating pattern.
This links to my current interest in deep time including the birth of the universe and emergence of complexity. The glass items also look wonderful when lit from rear. As such, I am considering mounting them over a light source. However, this remains to be seen…
In order to grow cells in the dishes, they need to be sterilised so that they do not carry any bacteria or other organisms that could contaminate my cells.
I am feeling more confident in using the benchtop autoclaves independently so am preparing a batch for sterilisation today. As per previous work, they are placed in autoclave bags and sealed with tape. Once the bags are autoclaved, black lines indicate successful sterilisation.
 Cut glass dish and wrapped dish ready for sterilisation.
Cut glass dish and wrapped dish ready for sterilisation.
I have also sources some small glass vials which I am considering integrating into some of the future creative works. There are various shapes that I am planning to test.
 Selection of glass vials for cell culture trial including metal closures.
Selection of glass vials for cell culture trial including metal closures.
 Glass vessels in autoclave bags ready for sterilisation.
Glass vessels in autoclave bags ready for sterilisation.
Finally, I have also prepared some additional 150mm and 90mm Petri dishes. The large dishes will be used as container vessels for the cut glass dishes to keep them sterile during cell culture.
 Petri dishes and other glass items in autoclave bags ready for sterilisation.
Petri dishes and other glass items in autoclave bags ready for sterilisation.
I divided the batch into two runs. As per previous process, I used cycle 6 (134 degrees for 10 min). This enables me to process both glassware and metal. It takes about 10 min for the sterilization process (but extra for cooling to handle materials).
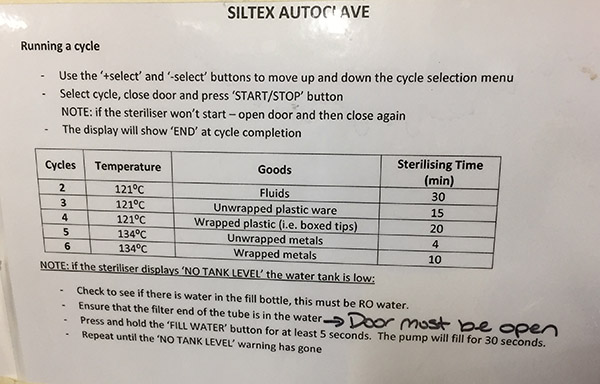 Autoclave instructions with cycle details.
Autoclave instructions with cycle details.
 Autoclaved bags containing sterilised Petri dishes.
Autoclaved bags containing sterilised Petri dishes.
 Autoclaved bags stored in lab area, ready for use.
Autoclaved bags stored in lab area, ready for use.