I am currently planning out my upcoming show (June 2022) at The Barracks. Michelle from the Derwent Valley Arts Committee indicated that the large courtyard would make a potentially interesting site to include in the show.
This prompted me to ponder how I might create a sculptural work suitable for an outdoor environment – and whether I could re-make a version of ‘Coming to Terms with Being Forgotten’ – but designed to disintegrate, shed nutrients and enable the growth of other plants and animals. In this way, the work could better reflect the idea of letting go and making way for what comes after. It would also force me to be more considerate about the materials used in the construction of the work.
 Svenja Kratz, Coming to Terms with Being Forgotten, 2020
Svenja Kratz, Coming to Terms with Being Forgotten, 2020
Fibreglass, polyurethane, plastic, plants, insects, wax, polymer clay, clay, acrylic paint. Original sculpture exhibited at Rosny Barn (2020) and The Barracks (2021).
This consideration prompted me to revisit the work of Australian artist Jamie North:
 Jamie North, Remainder No.4 2016
Jamie North, Remainder No.4 2016
cement, blast-furnace slag, coal ash, marble waste, living Australian plants
45cm diameter.
I like the impermanence disintegrating aesthetic and the integration of live plants into the structure.
Some of his larger works also hint at the ephemerality of all things including monuments and other markers of ‘human ingenuity’.
 Jamie North, Drifting to Void, 2016
Jamie North, Drifting to Void, 2016
Cement, blast-furnace slag, coal ash, marble waste, steel, living Australian plants, 240 x 67 x 67cm
Jamie North’s work also connects to the ‘TerraForm’ sculptures of Robert Cannon – although I must admit that I prefer the more abstracted works.
 Robert Cannon, Apollo, concrete, moss and living plants via: Design Swan
Robert Cannon, Apollo, concrete, moss and living plants via: Design Swan
 Robert Cannon, Uprising, concrete, moss and living plants via: Design Swan
Robert Cannon, Uprising, concrete, moss and living plants via: Design Swan
The work of Antony Gormley is also always interesting to consider in relation to the human figure.
In the context of this project, I think the work ‘Sense’ offers an interesting option in relation to the idea of an absent body (with the potential of filling of a void with potential growth).
 Antony Gormley, Sense, 1991, concrete
Antony Gormley, Sense, 1991, concrete
While concrete is one material option for producing an outdoor sculpture, I really want to find an alternative casting material that would offer better biodegradability alongside nutrient supplements for organism growth and soil improvement.
There seem to be a growing number of more sustainable materials or biodegradable materials available for casting or injection moulding including bioplastics or Arboform, which manufacturers describe as ‘Liquid Wood’. However, a number of these products seem to be more designed for industrial and product design purposes. As such, I think this project is more suited to raw materials such as compost or a custom mix between a variety of elements (e.g. sand, rocks, compost, concrete, hay) to enable a range of durational unfoldings and both nutrients and potential habitats.
To help with some of the planning, I consulted designer (and UTAS concrete guru) Jouni Jarvela. He suggested that casting would be a good option, but it would be best to test a range of materials on a smaller scale before sizing the design up to a large-scale format. I do love me some design prototyping!
Following our chat, I will work on a smaller ‘bust’ version which will be easier to manage than a life-size human form. In the interim, he will consider some material options for testing.
I have to say that one of the things I love about making work is seeing where things go after an initial idea is put forward. At this stage, a life-size courtyard sculpture for June seems out of the realm of possibility – however, a series of degrading self-portrait busts may be the alternative outcome. Who knows….
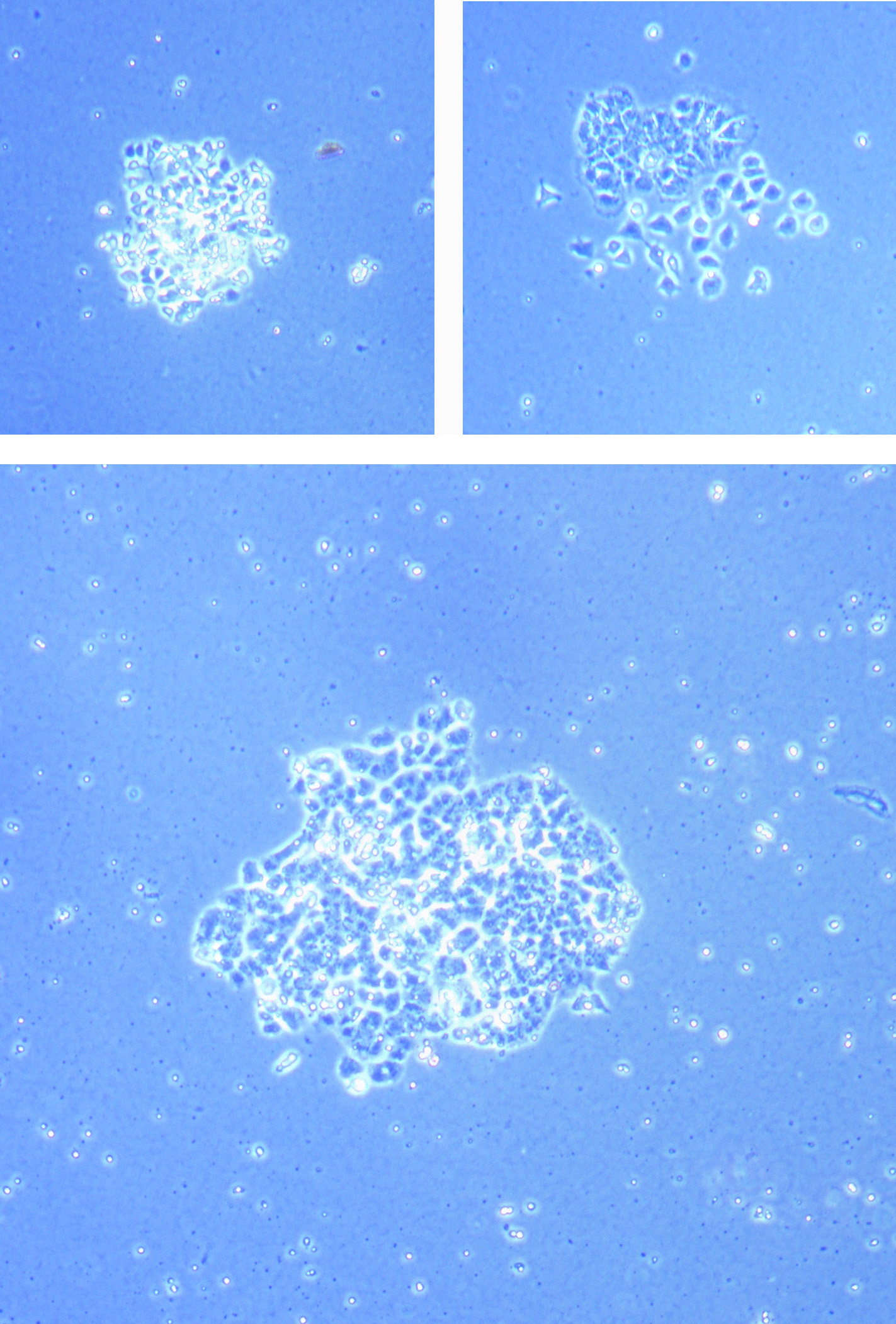
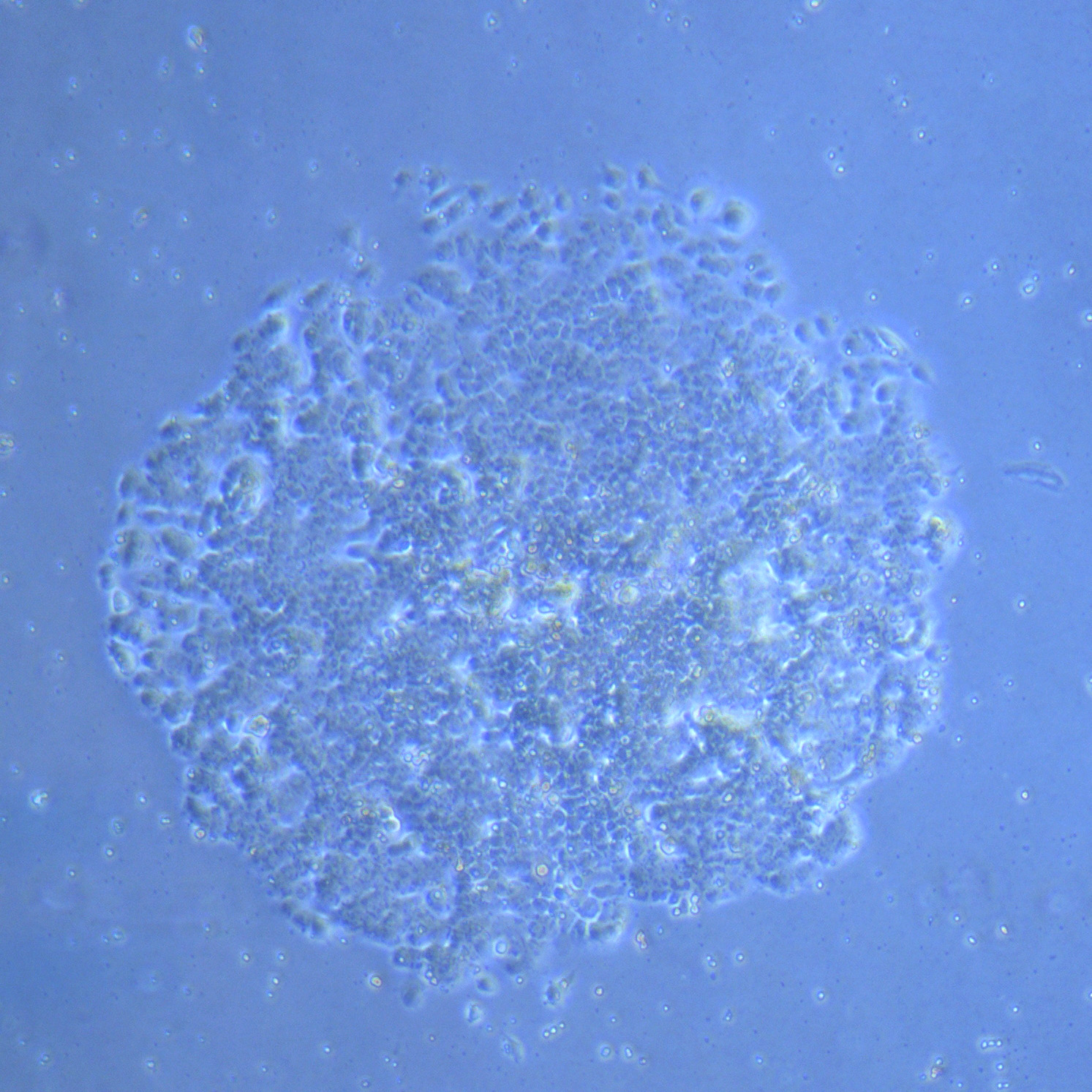
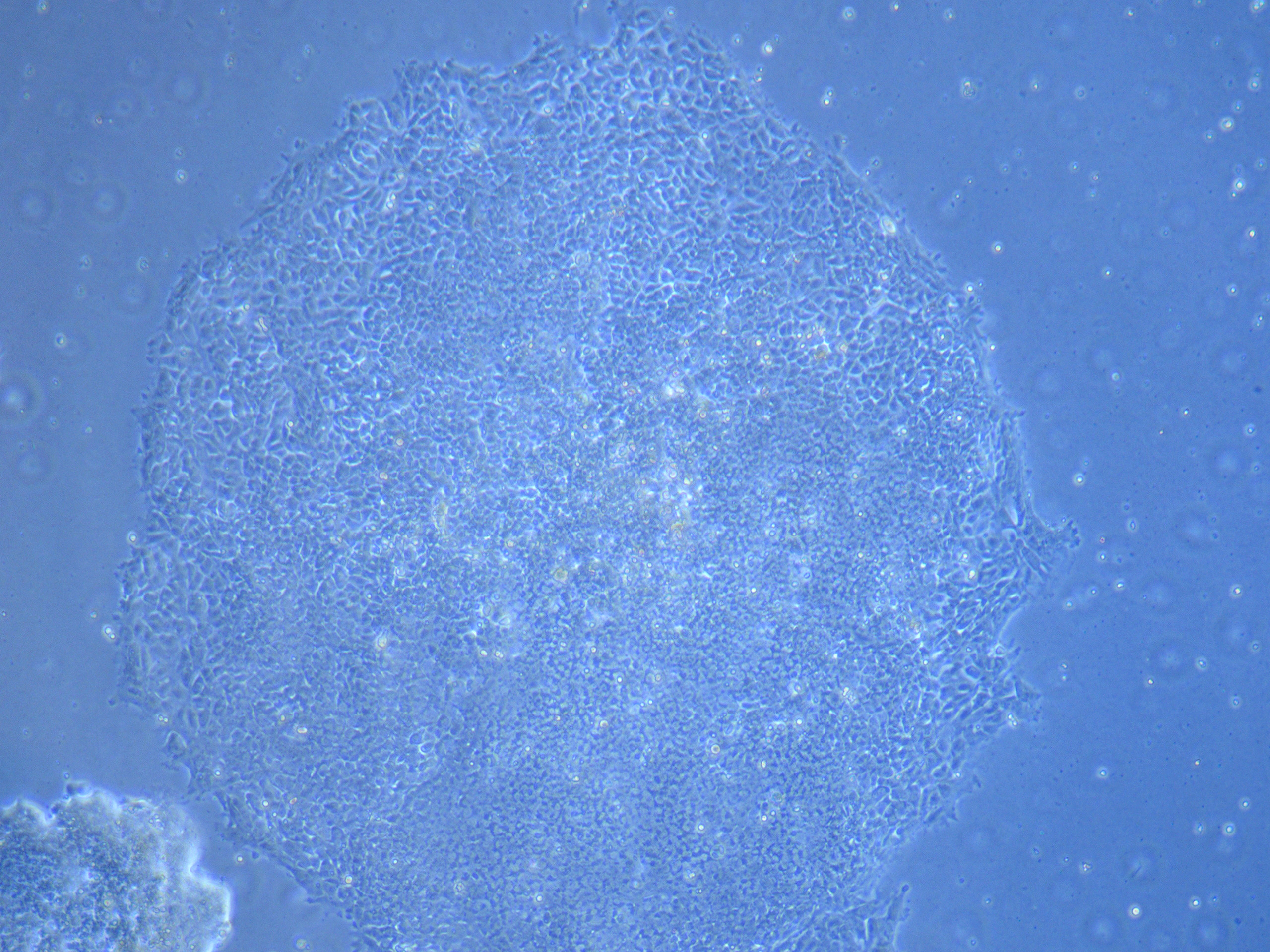
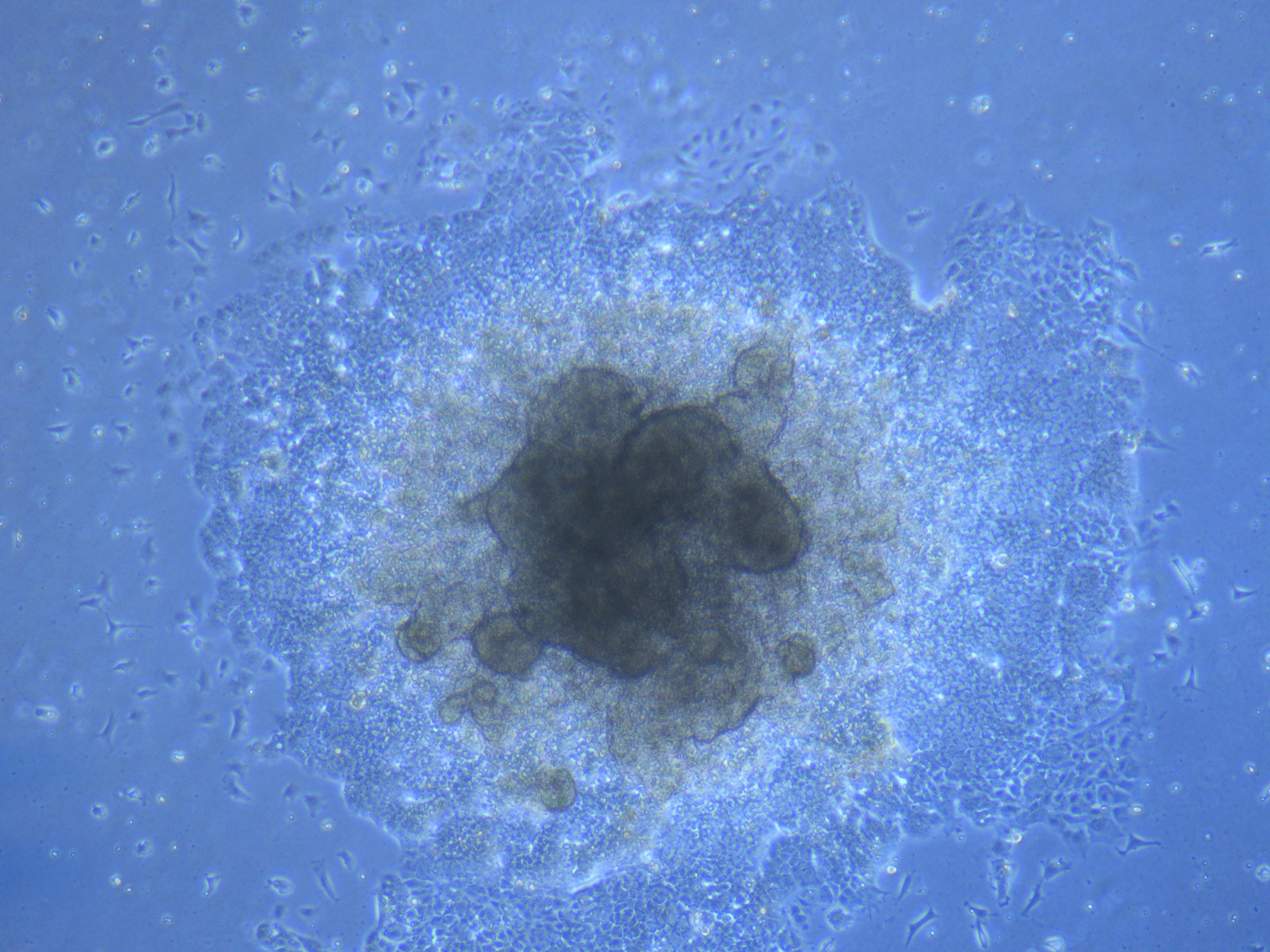
 Light microscope image of cut glass dish with media containing dead cell debris and evidence of a small number of surviving cells.
Light microscope image of cut glass dish with media containing dead cell debris and evidence of a small number of surviving cells. 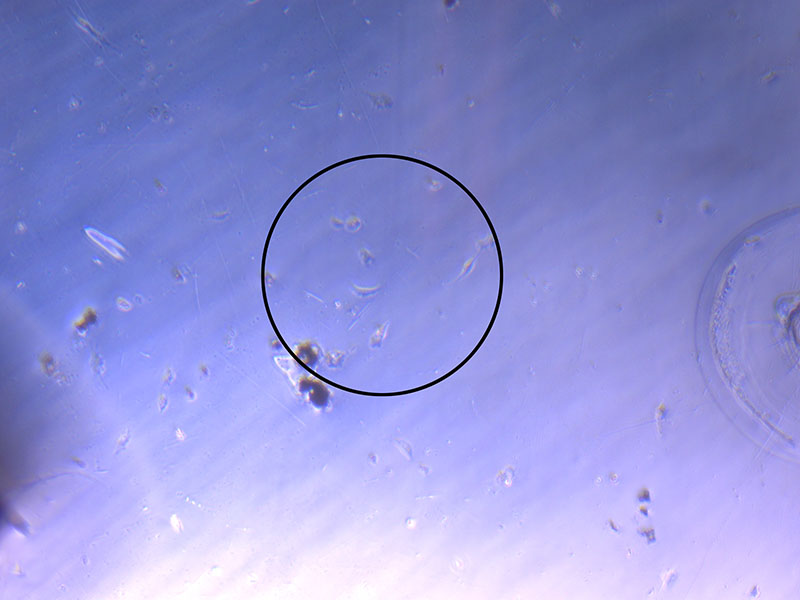
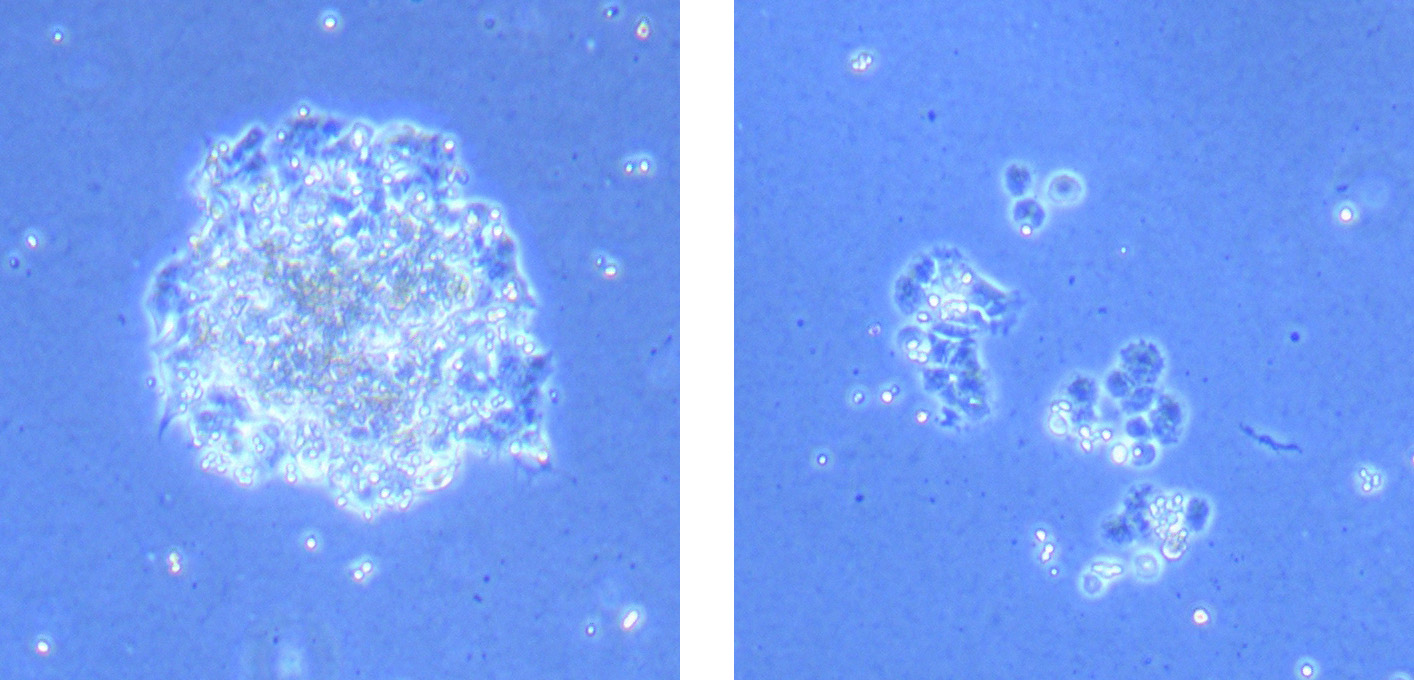
 Light microscope image of cut glass dish in PBS showing further evidence of a small number of surviving cells.
Light microscope image of cut glass dish in PBS showing further evidence of a small number of surviving cells.  Light microscope image of cut glass dish in PBS with likely cells circled. There are a few additional potential cells visible, but I have only circled the most obvious.
Light microscope image of cut glass dish in PBS with likely cells circled. There are a few additional potential cells visible, but I have only circled the most obvious. 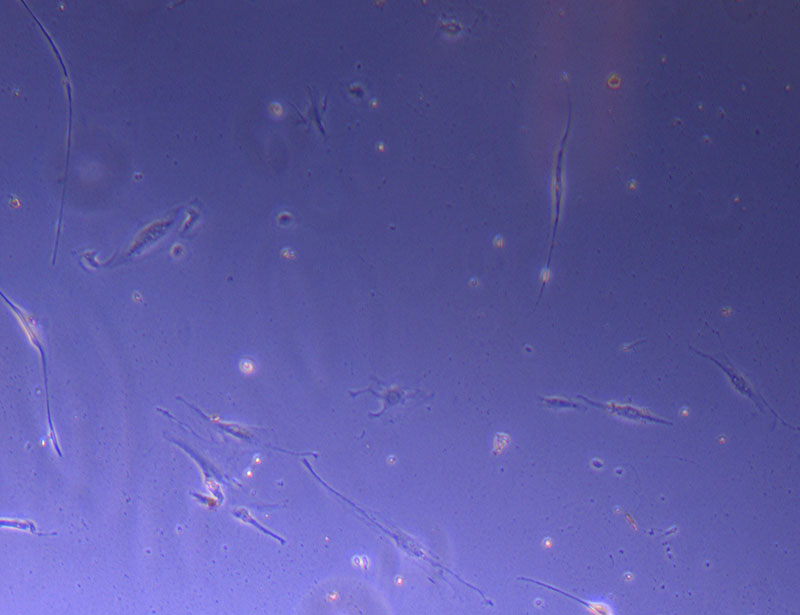 Light microscope image of Petri dish with fresh complete media with live cells.
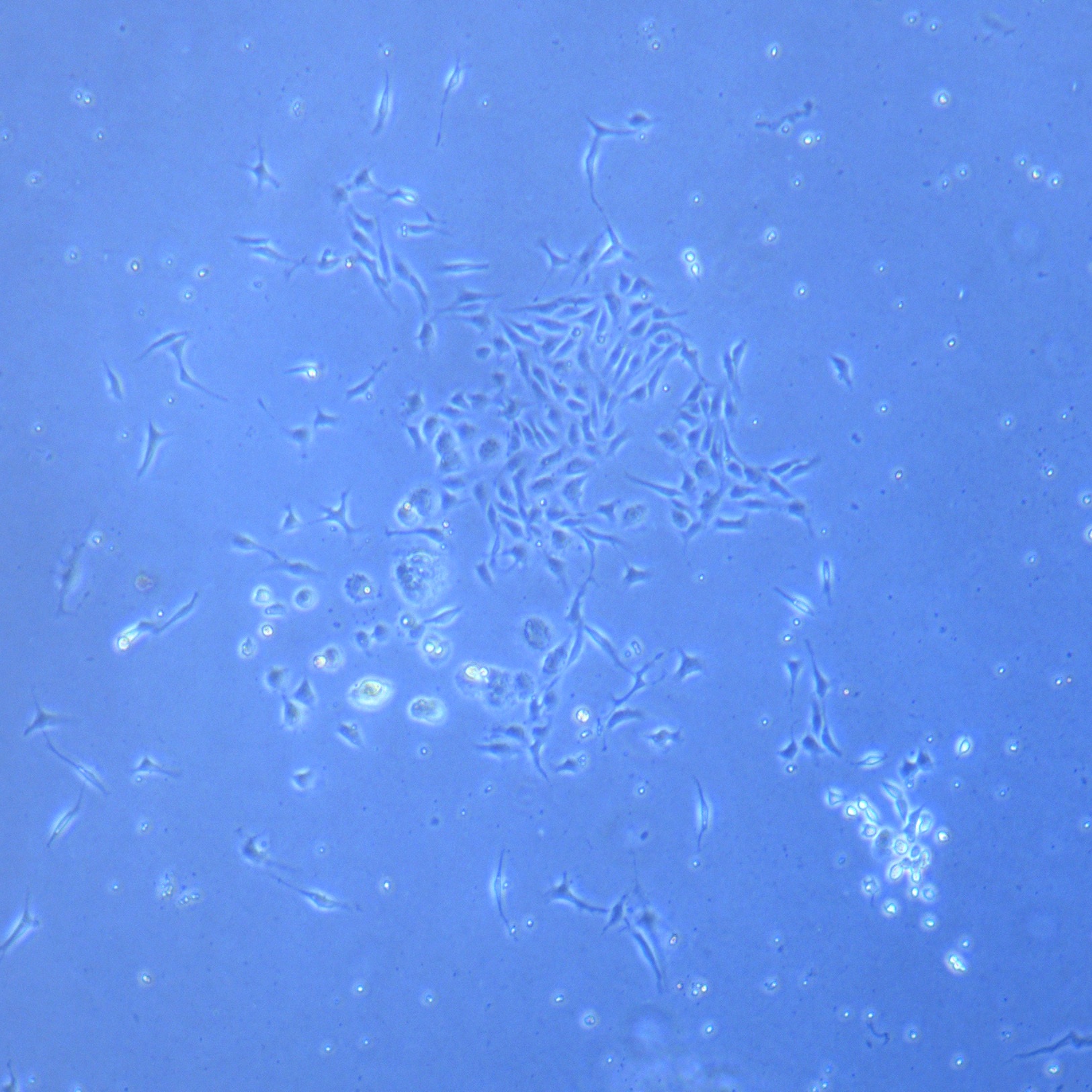
Light microscope image of Petri dish with fresh complete media with live cells. 
 Simple graphic interface with presets ready to complete fluorescent microscopy.
Simple graphic interface with presets ready to complete fluorescent microscopy. 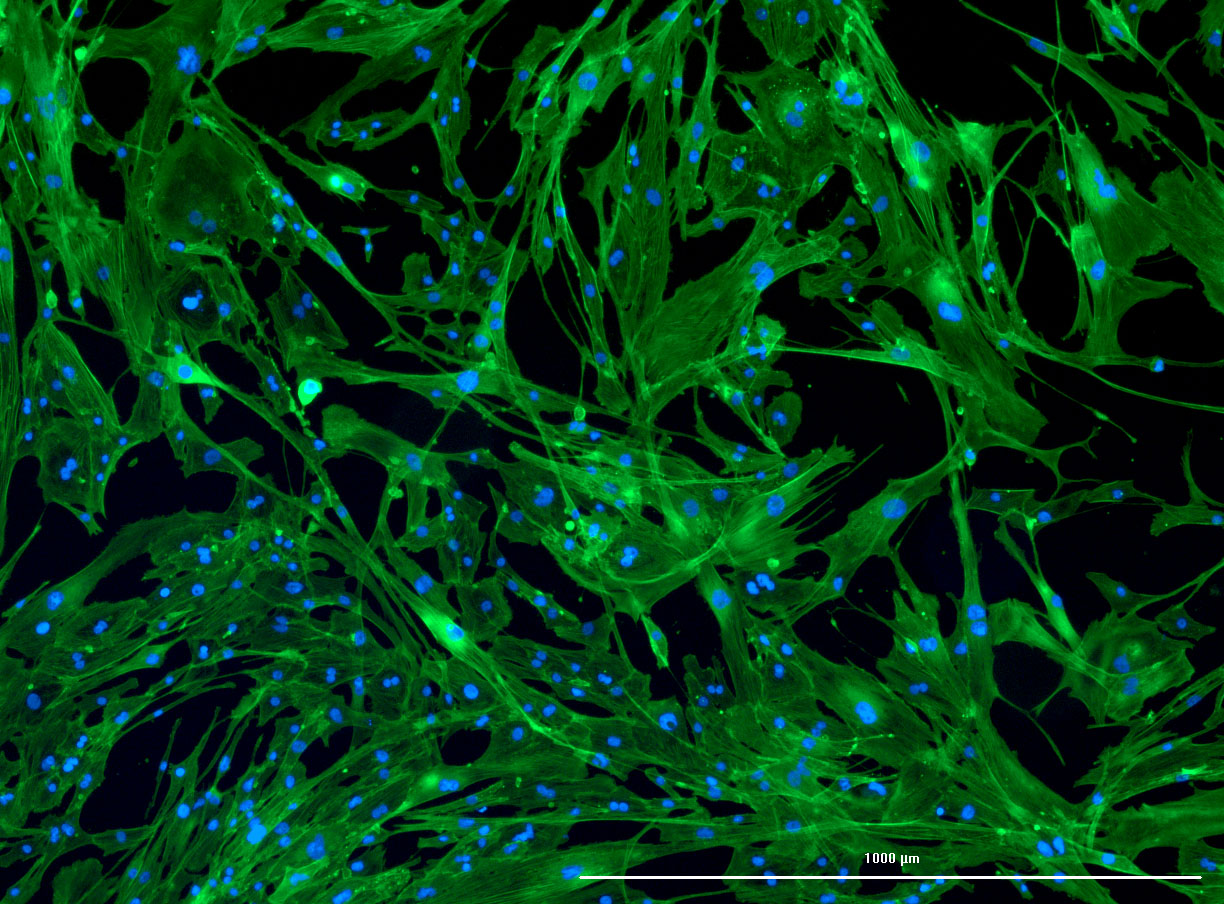
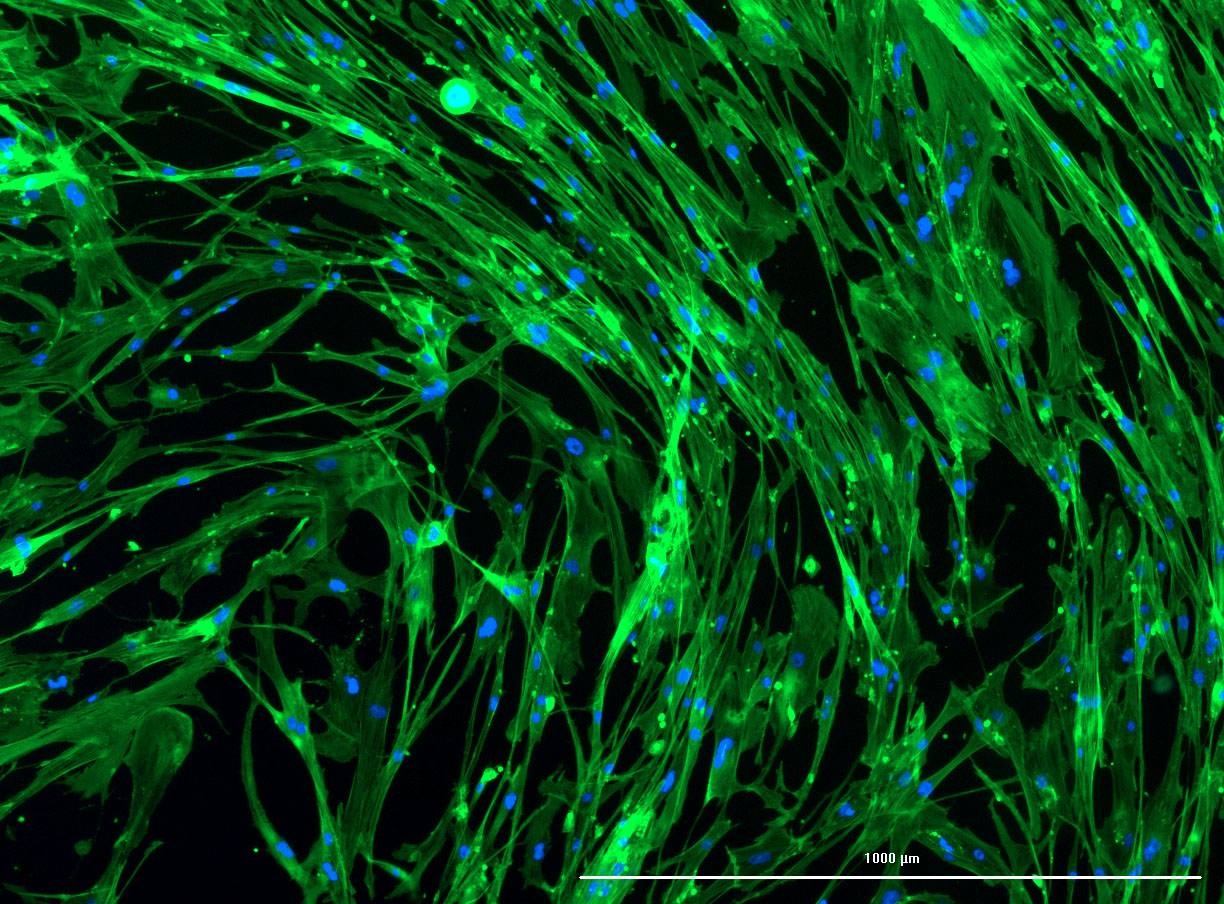
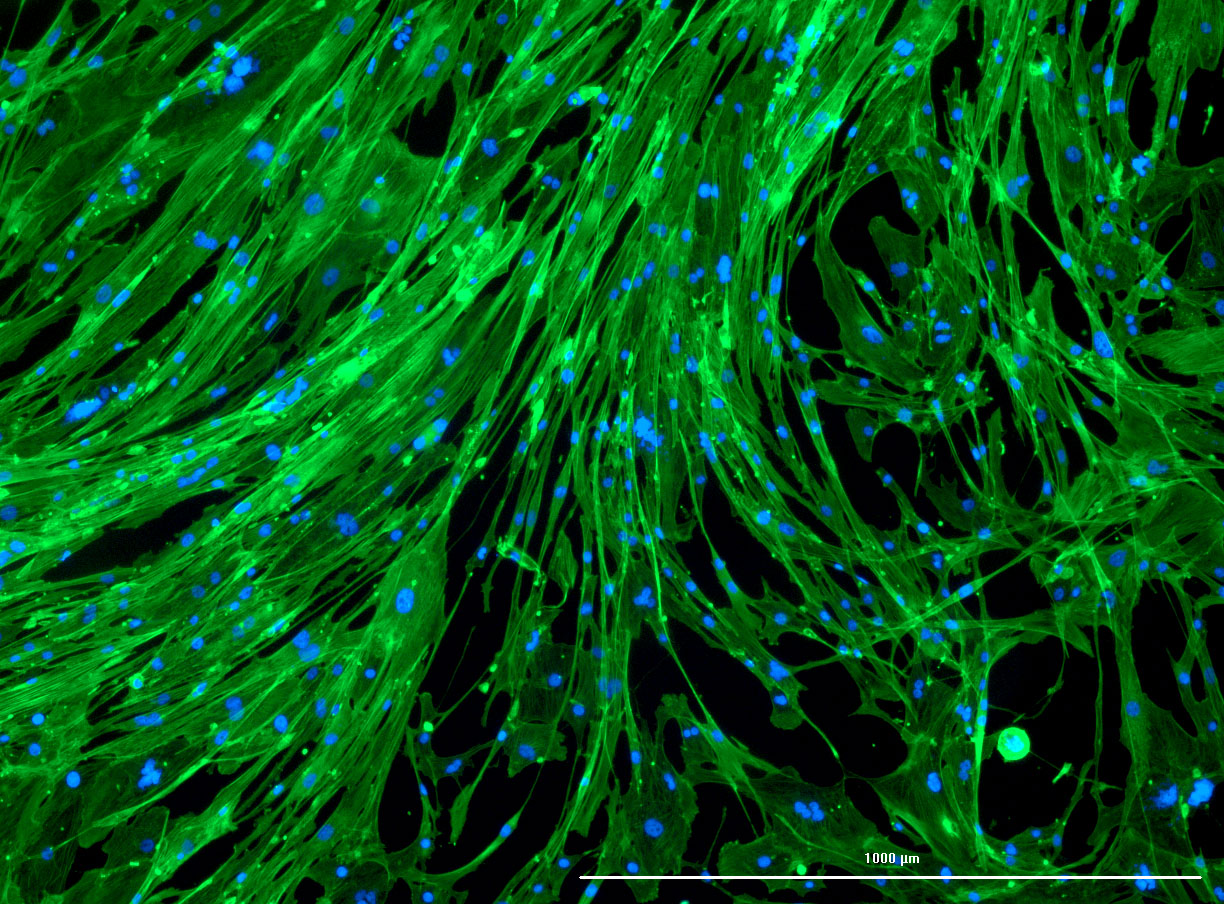
 DAPI and Fluorescein Phalloidin staining of confluent fibroid cells P4 (although this is potentially misleading as the cells are very slow growing).
DAPI and Fluorescein Phalloidin staining of confluent fibroid cells P4 (although this is potentially misleading as the cells are very slow growing). 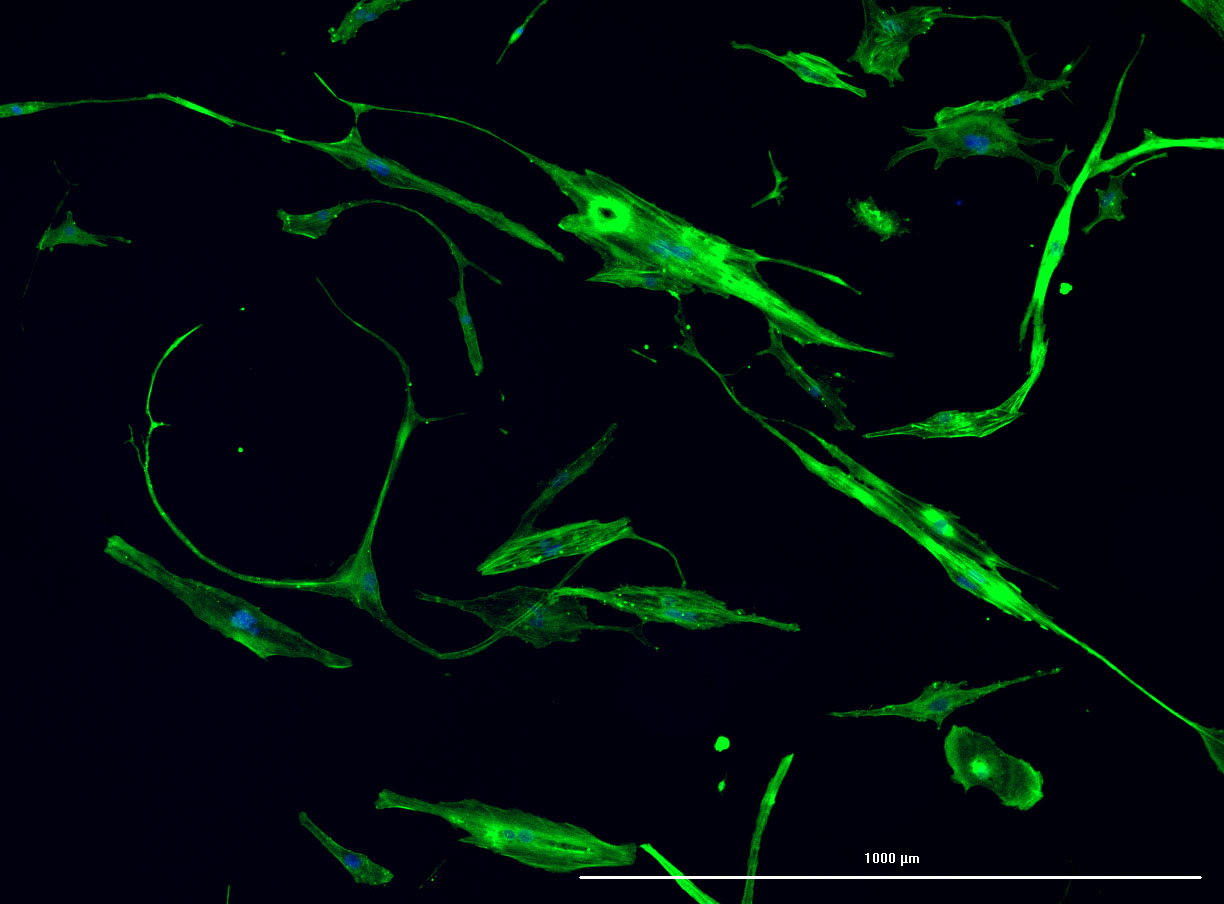
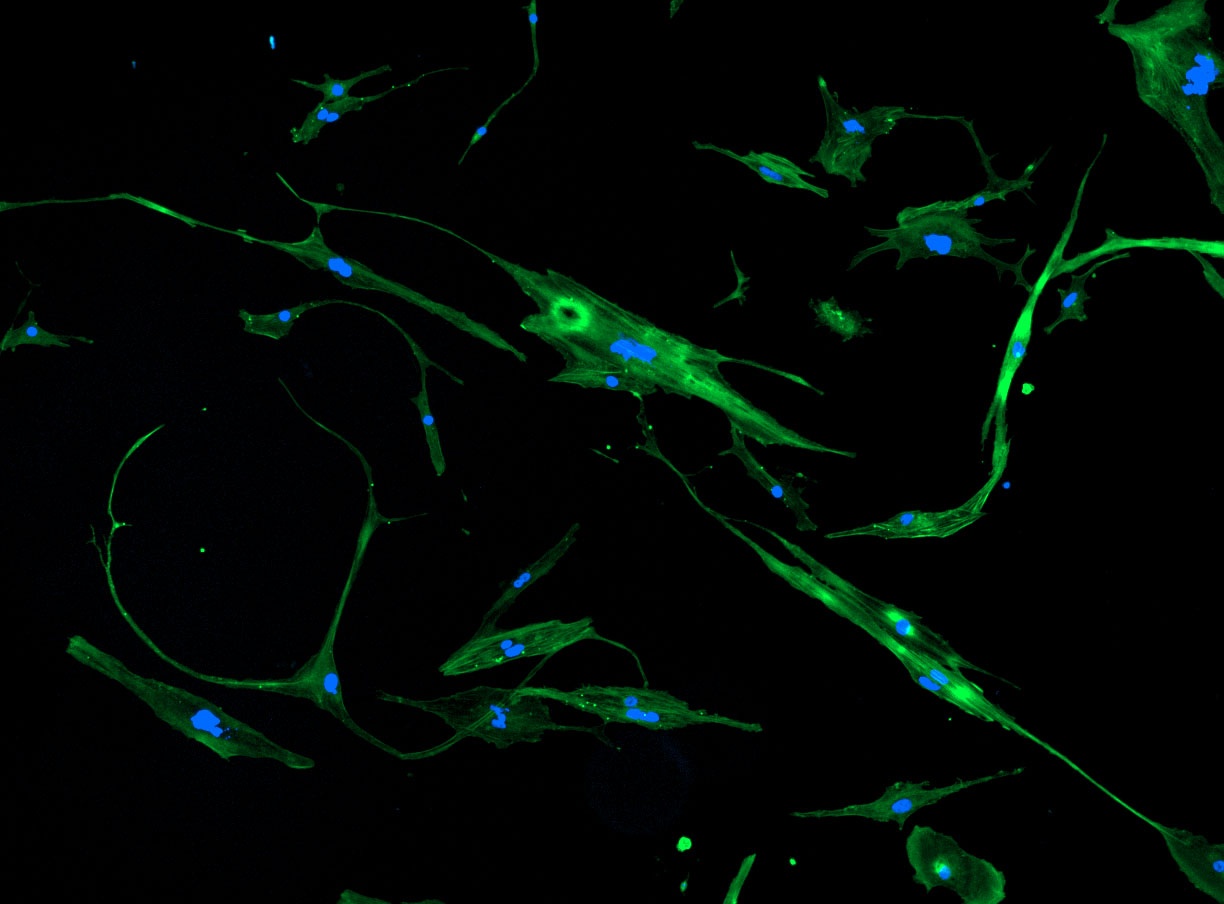
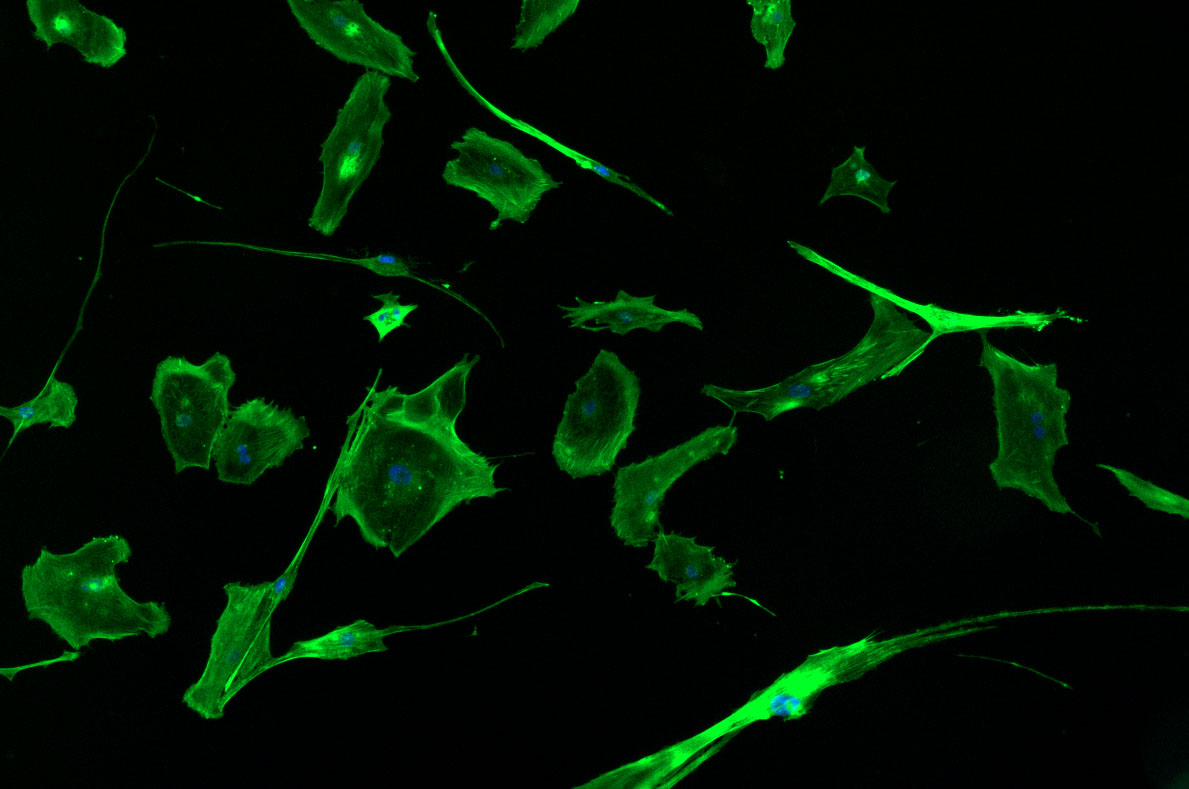
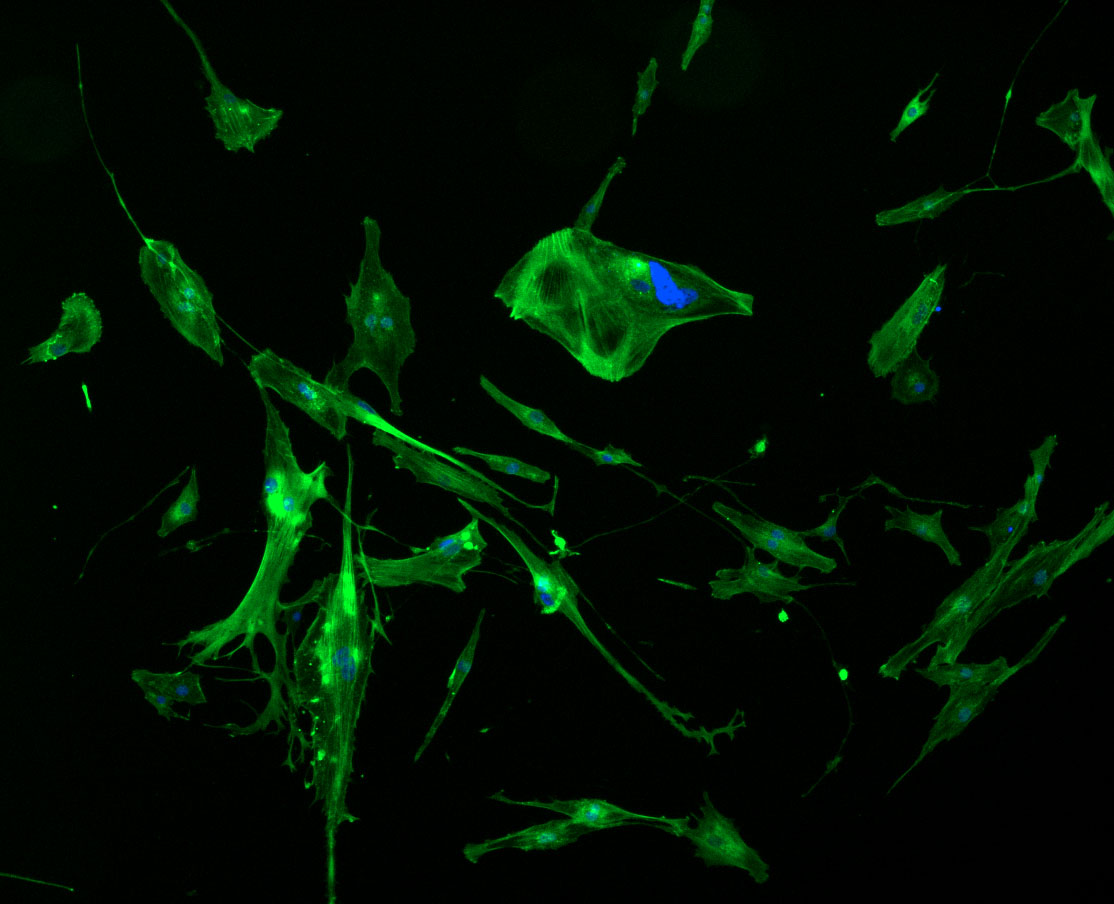
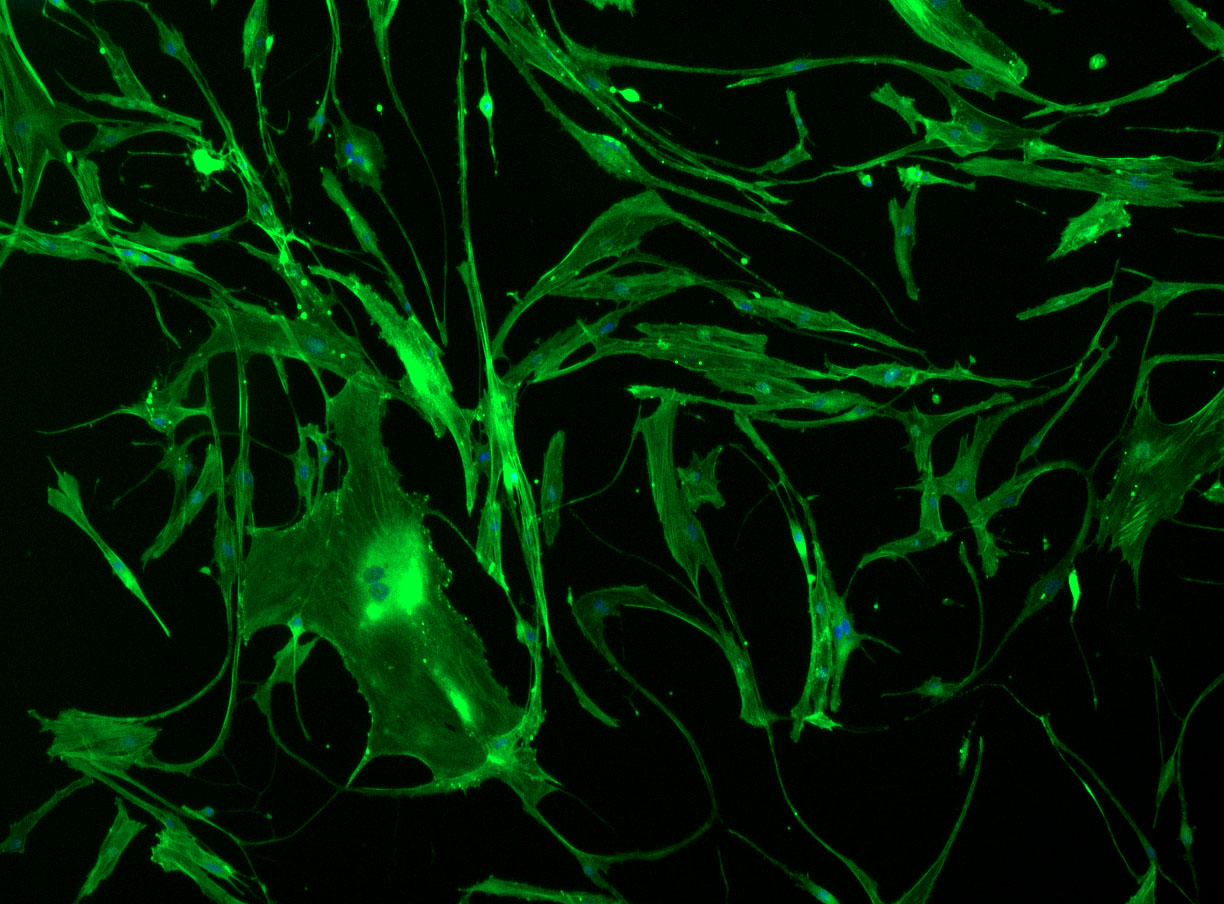
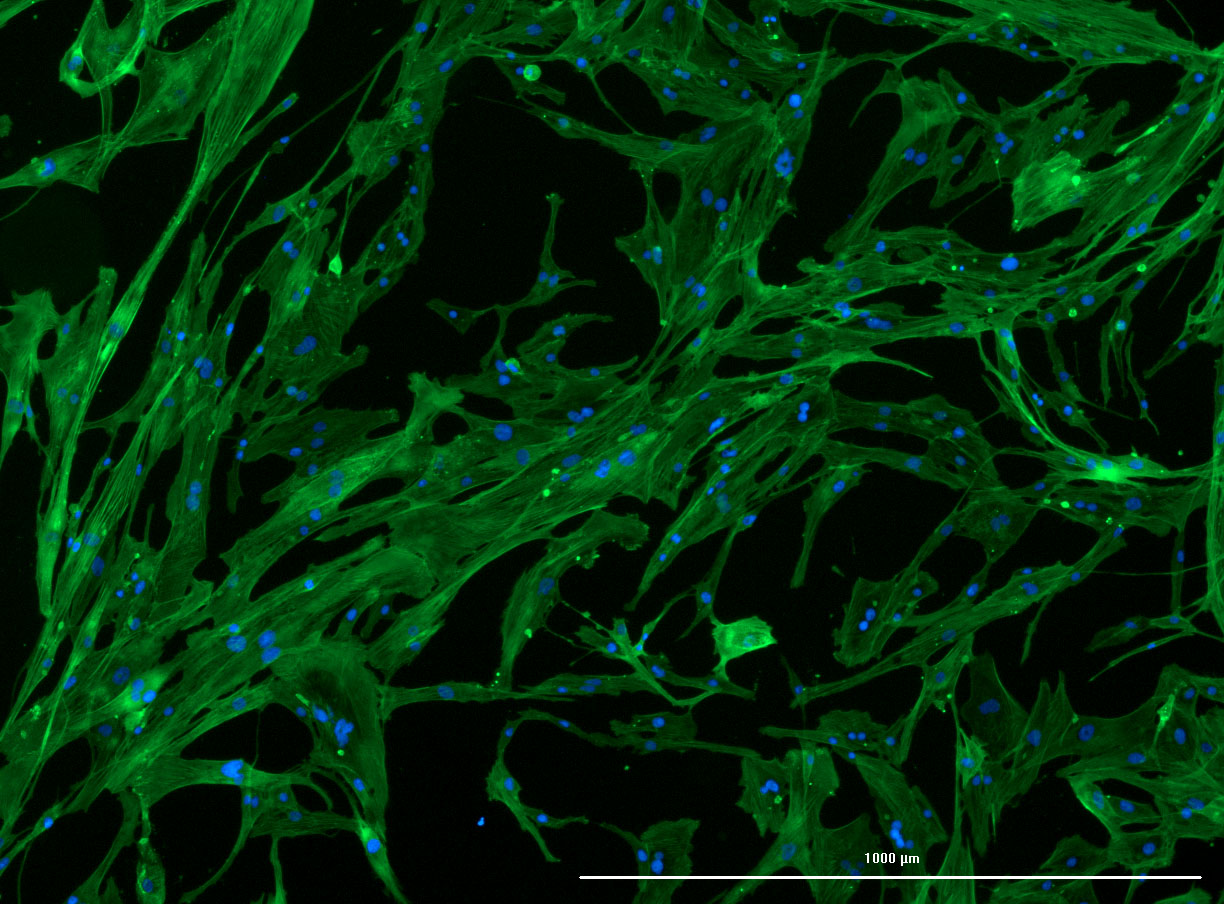
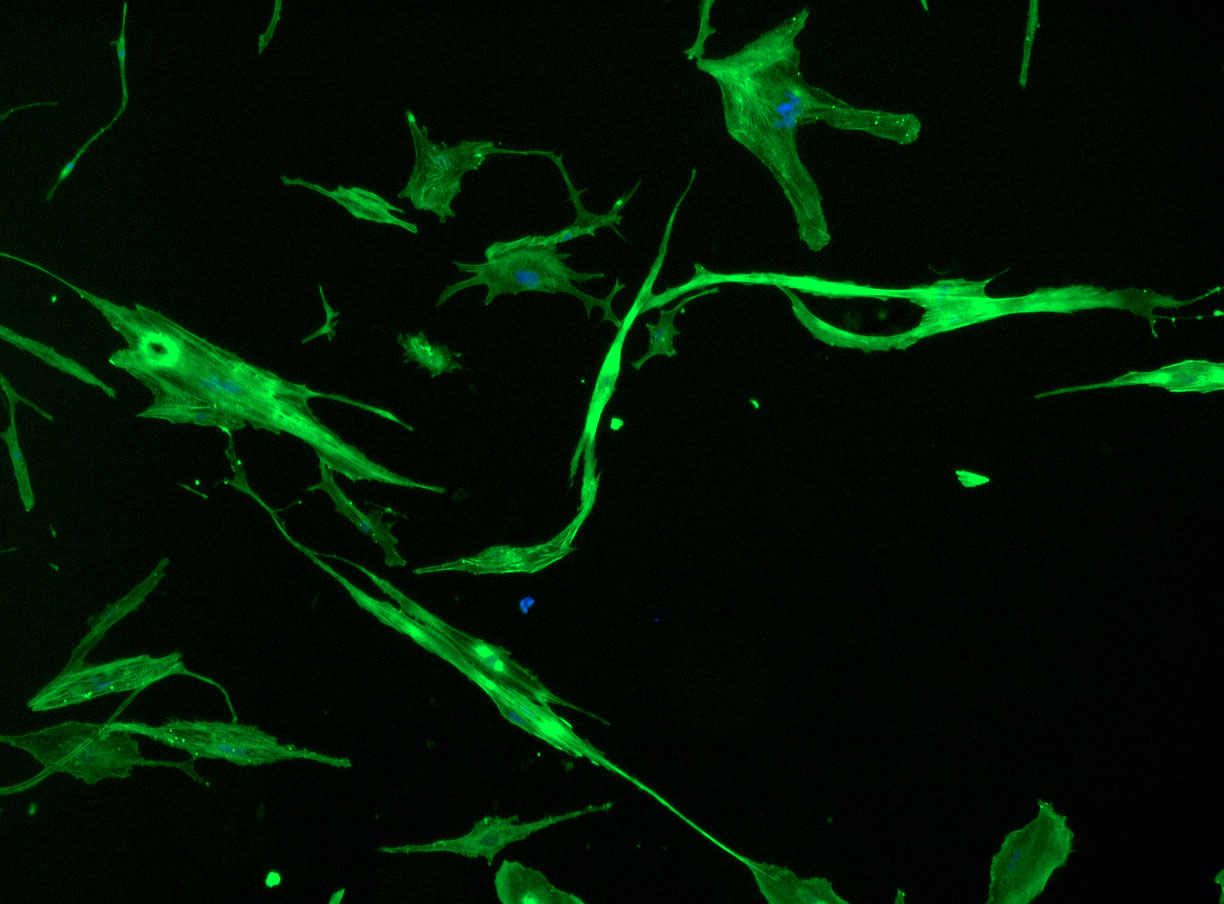 DAPI and Fluorescein Phalloidin staining of fibroid cells P4 which enables better visualisation of individual cells.
DAPI and Fluorescein Phalloidin staining of fibroid cells P4 which enables better visualisation of individual cells.  Working with 4% PFA in Fume Hood
Working with 4% PFA in Fume Hood Working at lab bench in the Stroke Group area
Working at lab bench in the Stroke Group area

 Unwrapping Petri dishes and getting ready to coat culture glassware with PLL.
Unwrapping Petri dishes and getting ready to coat culture glassware with PLL.  PLL coated glassware in Petri dishes ready for incubation.
PLL coated glassware in Petri dishes ready for incubation.  Cut glass vessels with cells ready for incubation.
Cut glass vessels with cells ready for incubation. 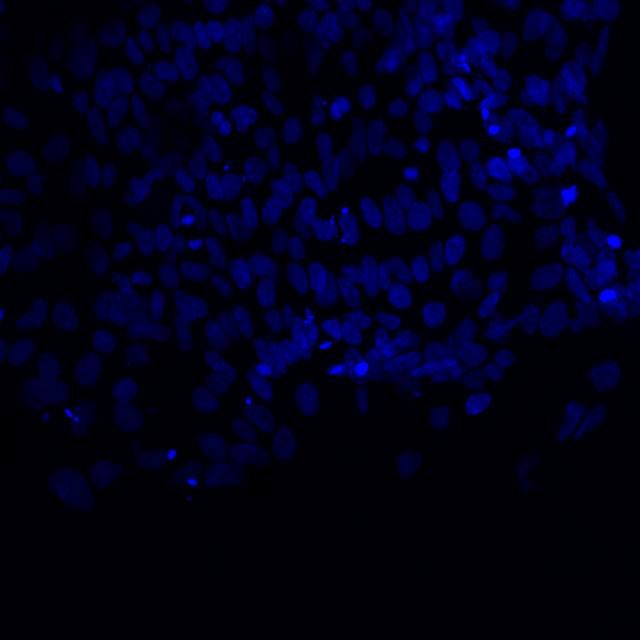 Microscopic image of stem cells, Hues 9 stained with DAPI (blue) by the UC San Diego Stem Cell Program.
Microscopic image of stem cells, Hues 9 stained with DAPI (blue) by the UC San Diego Stem Cell Program. Image of Amanita phalloides via
Image of Amanita phalloides via 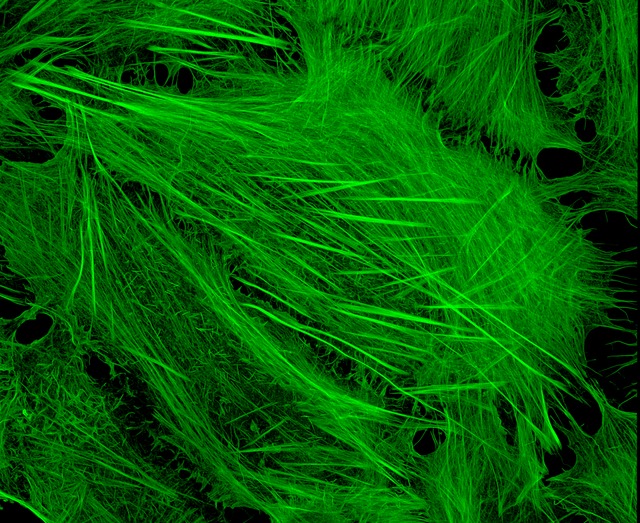 U2OS cells stained with fluorescent phalloidin taken on a confocal microscope by Howard Vindin
U2OS cells stained with fluorescent phalloidin taken on a confocal microscope by Howard Vindin{kind=link}