While I wait for ethics clearance and team members to return to the lab, I am continuing to culture my cells.
3 x T75 and 1 x Cut Glass Dish ready for cell maintenance (media change) on 21/1/22.
At this point, I am feeding more confluent cells on a weekly basis (depending on how they look):
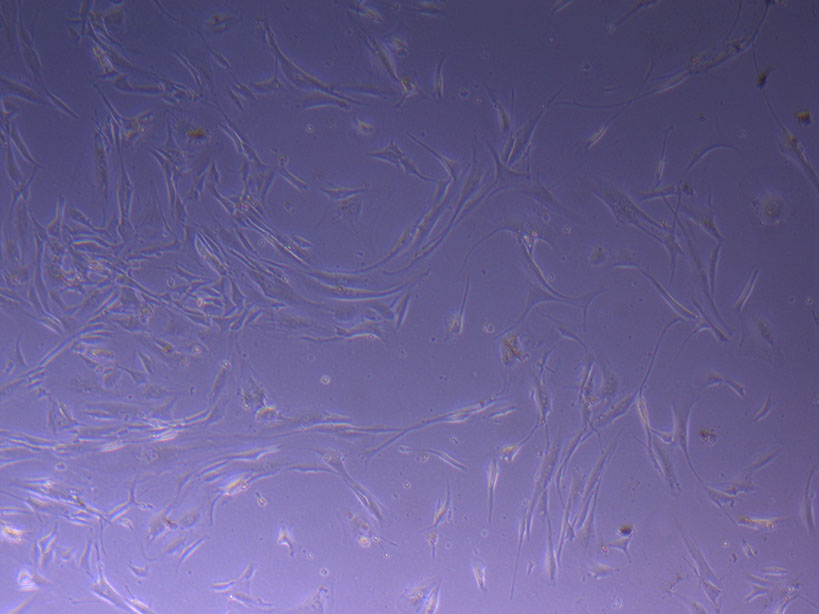 T75 originally plated 15/12/21 – Flask #1
T75 originally plated 15/12/21 – Flask #1
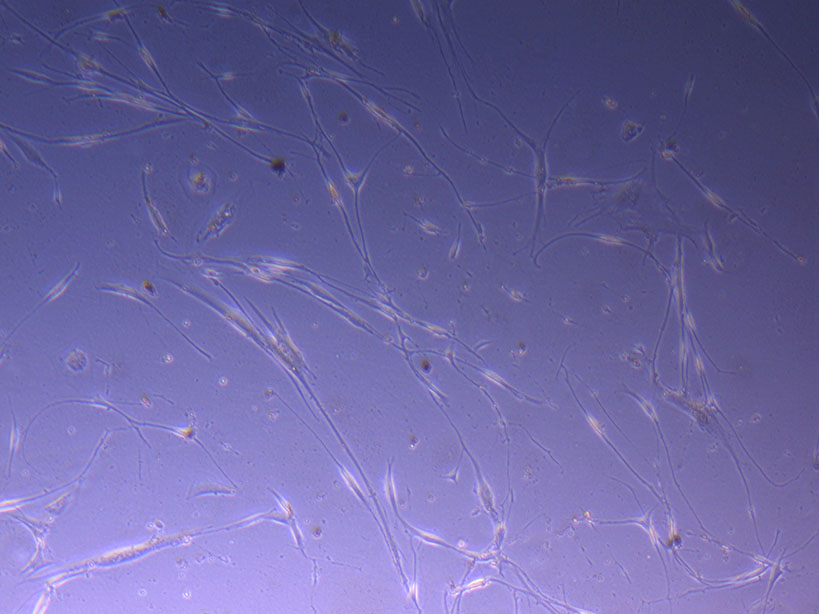 T75 originally plated 15/12/21 – Flask #2
T75 originally plated 15/12/21 – Flask #2
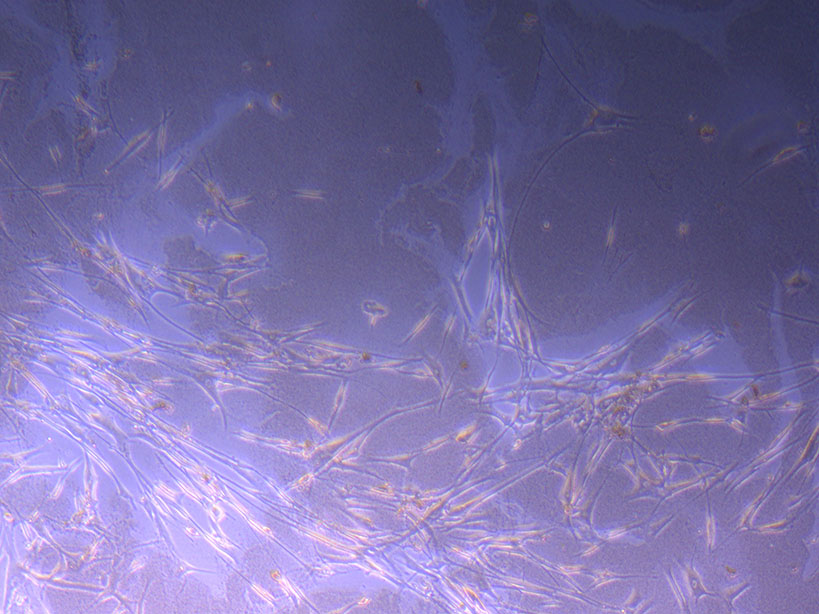 T75 originally plated 7/10/21
T75 originally plated 7/10/21
 Cut Glass Dish – originally plated 15/12/21
Cut Glass Dish – originally plated 15/12/21
While the cut glass dish is hard to image due to the more uneven surface of the glass, and being contained in a larger Petri Dish, there seems to be a good level of cell growth. Perhaps this will result in better staining in the next round.
I still have four other T75s. However, since these are less dense in cell numbers, I am limiting media change to reduce ongoing maintenance costs and media usage:
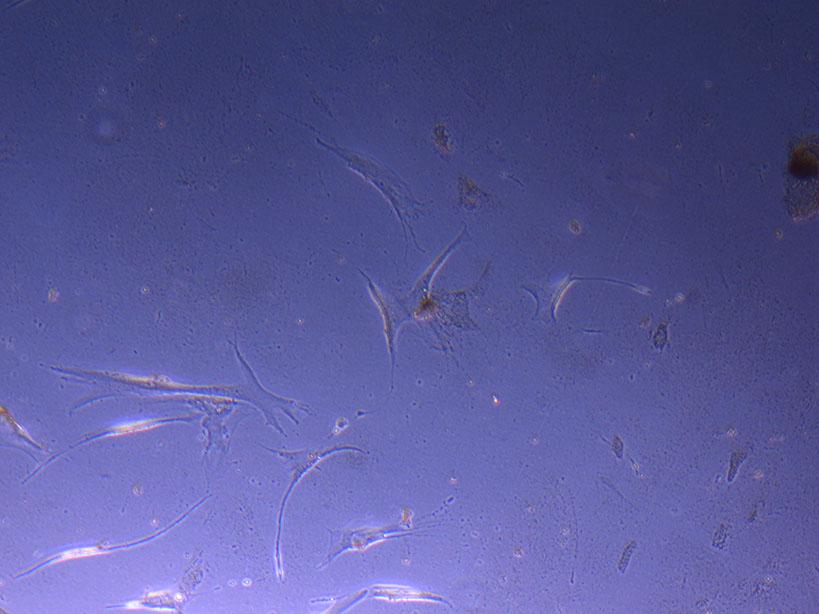 T75 originally plated 7/10/21 and passaged 16/11/21 – Flask #1
T75 originally plated 7/10/21 and passaged 16/11/21 – Flask #1
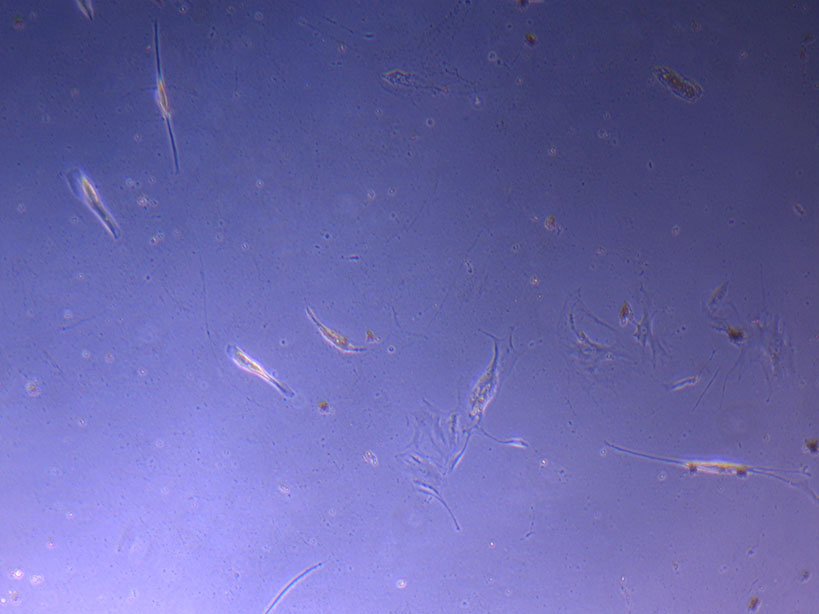 T75 originally plated 7/10/21 and passaged 16/11/21 – Flask #2
T75 originally plated 7/10/21 and passaged 16/11/21 – Flask #2
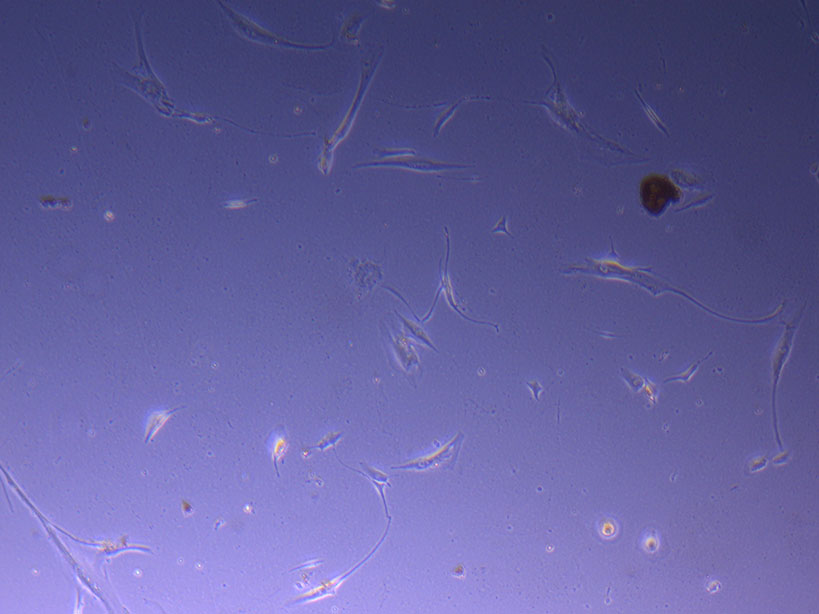 T75 originally plated 15/12/21 – Flask #3
T75 originally plated 15/12/21 – Flask #3
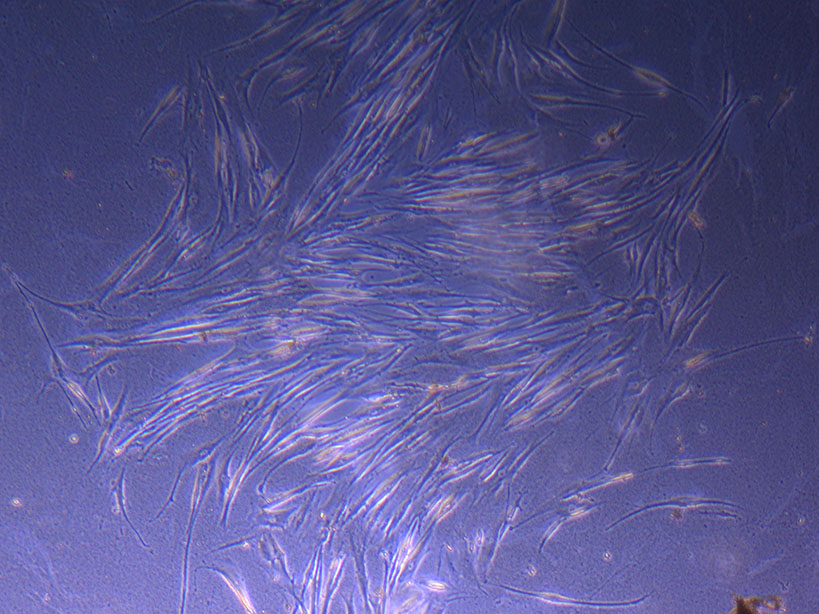
I must admit that I am continually amazed that the original flask of cells plated by Jo-Maree in August last year (and passaged a month later) still has viable cells in it. While much of the T75 flask area is pretty sparse with cells, there are some larger clusters including a growing bunch of elongated fibroid-like cells:
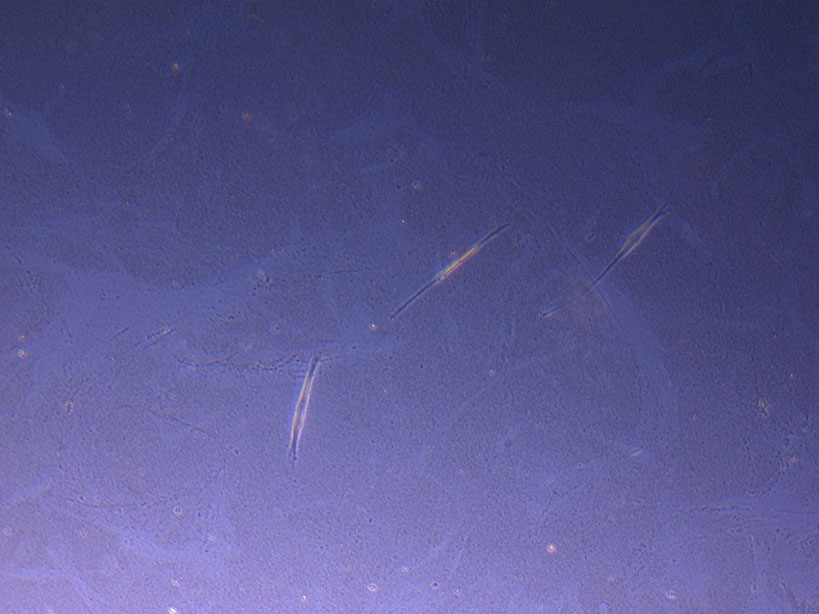 T75 originally plated 10/09/21 showing sparse number of cells with evidence of previous cell movement and presence in ‘ghost trails’.
T75 originally plated 10/09/21 showing sparse number of cells with evidence of previous cell movement and presence in ‘ghost trails’.
 T75 originally plated 10/09/21 showing small cluster of cells with evidence of previous cell movement and presence in ‘ghost trails’.
T75 originally plated 10/09/21 showing small cluster of cells with evidence of previous cell movement and presence in ‘ghost trails’.
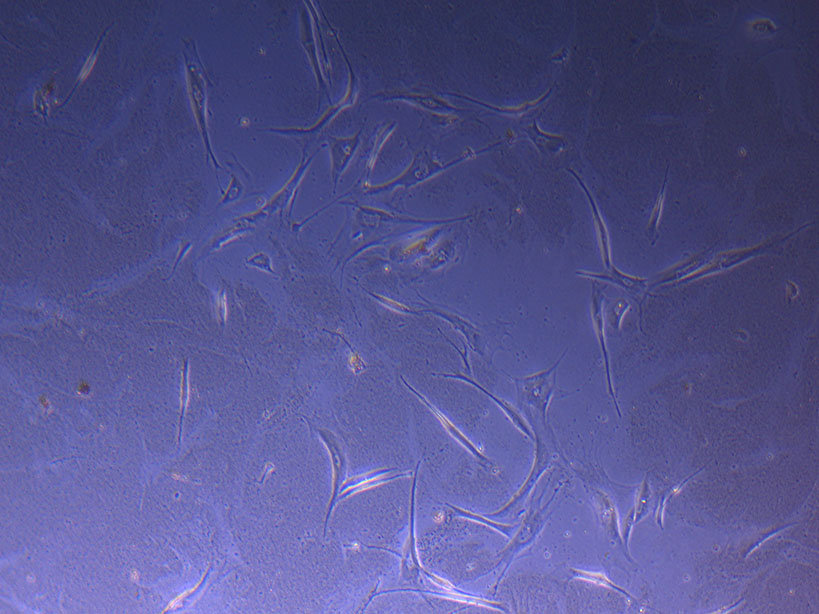
 T75 originally plated 10/09/21 showing larger cluster of fibroid-like cells.
T75 originally plated 10/09/21 showing larger cluster of fibroid-like cells.
They are easy to miss, but I’ve made a note to monitor their progress and see how they proliferate. Again…it just takes one mutant to get a cell line going!
 Image of PHGL Tumour Baby cells Flask with original plating date (P 3, 07/10/21) and passage date (P4, 09/11/21) recorded.
Image of PHGL Tumour Baby cells Flask with original plating date (P 3, 07/10/21) and passage date (P4, 09/11/21) recorded. 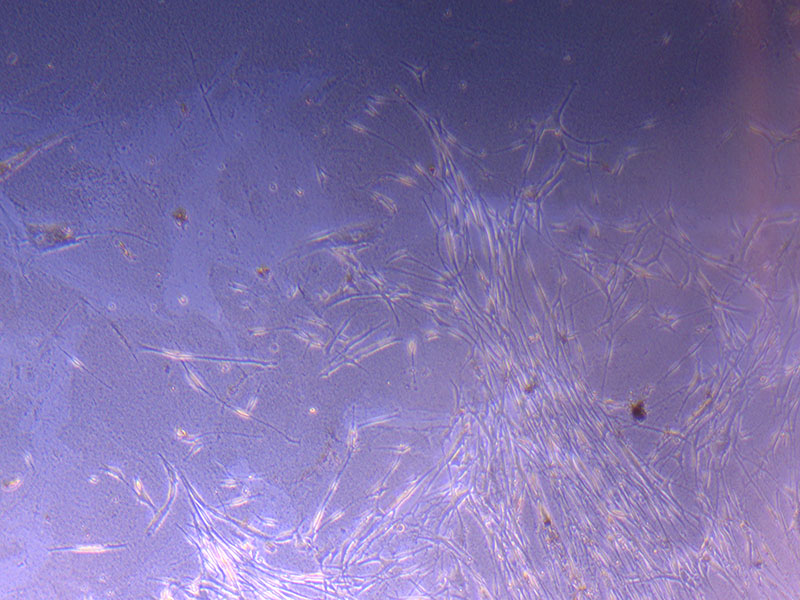 Light microscope image of PHGL Tumour Baby P3 in T75 flask [Org plated 07/10/21] imaged on 15/12/21.
Light microscope image of PHGL Tumour Baby P3 in T75 flask [Org plated 07/10/21] imaged on 15/12/21.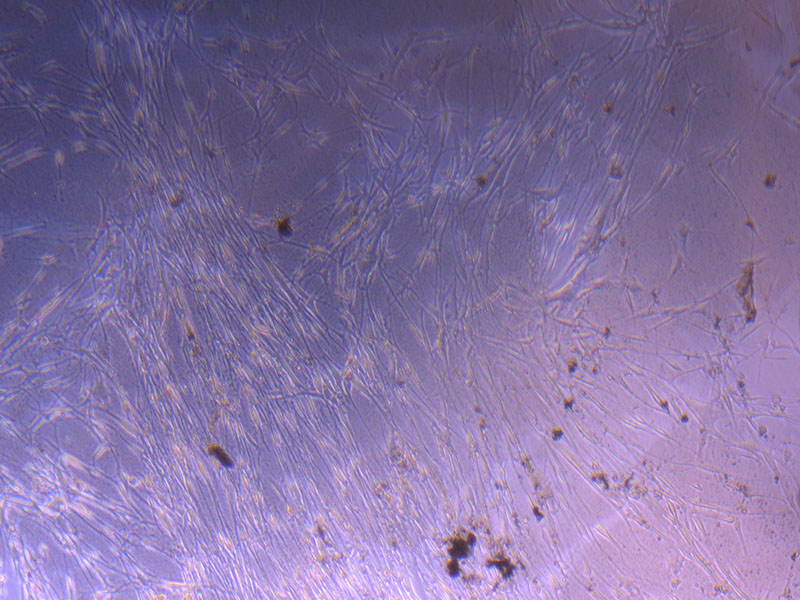
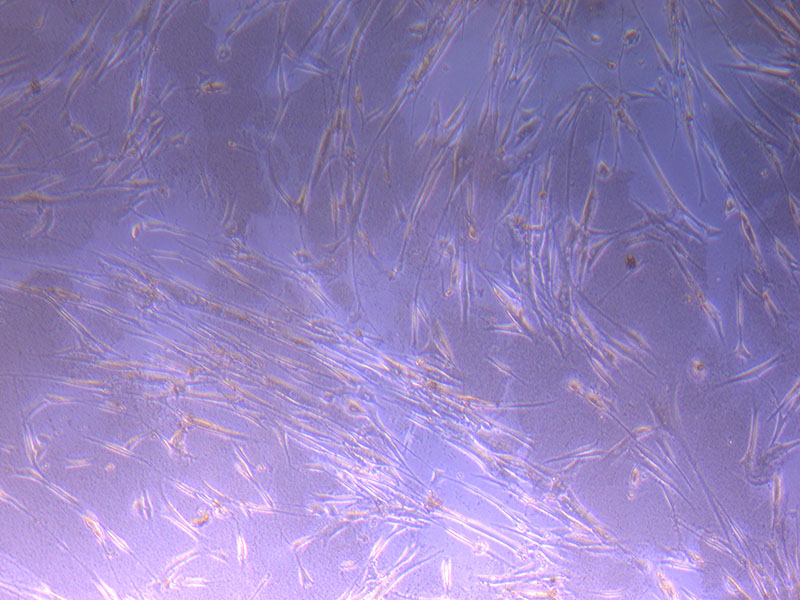
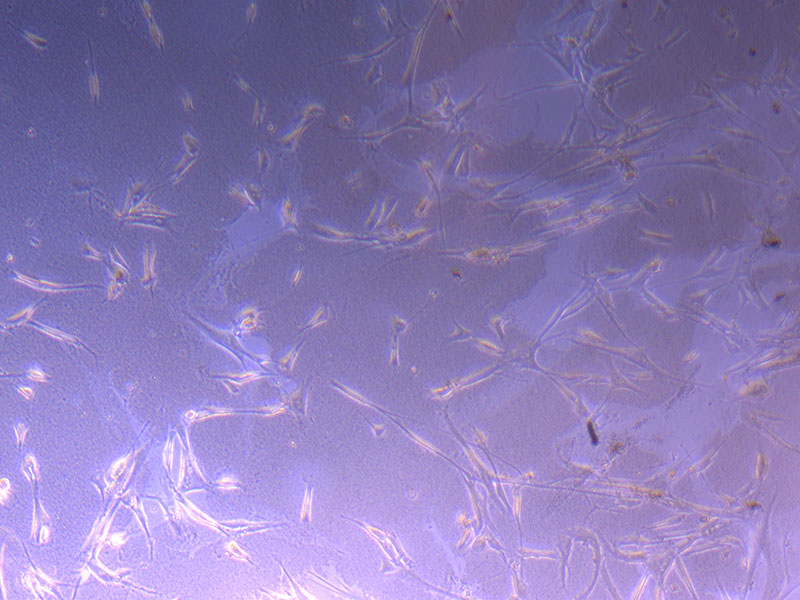
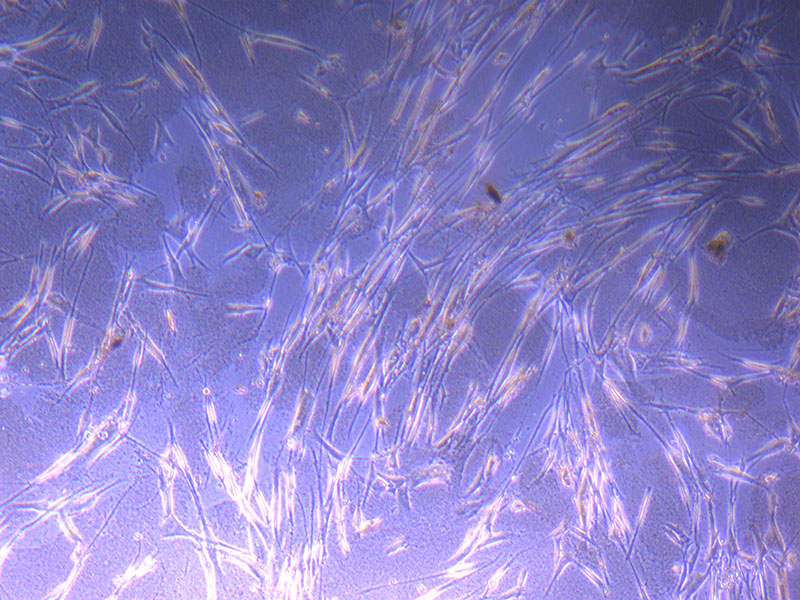 Light microscope images of PHGL Tumour Baby P4 in T75 flask [Org P3 plated 07/10/21 – passaged P4 on 9/11/21] imaged on 13/1/22 after cell maintenance.
Light microscope images of PHGL Tumour Baby P4 in T75 flask [Org P3 plated 07/10/21 – passaged P4 on 9/11/21] imaged on 13/1/22 after cell maintenance. 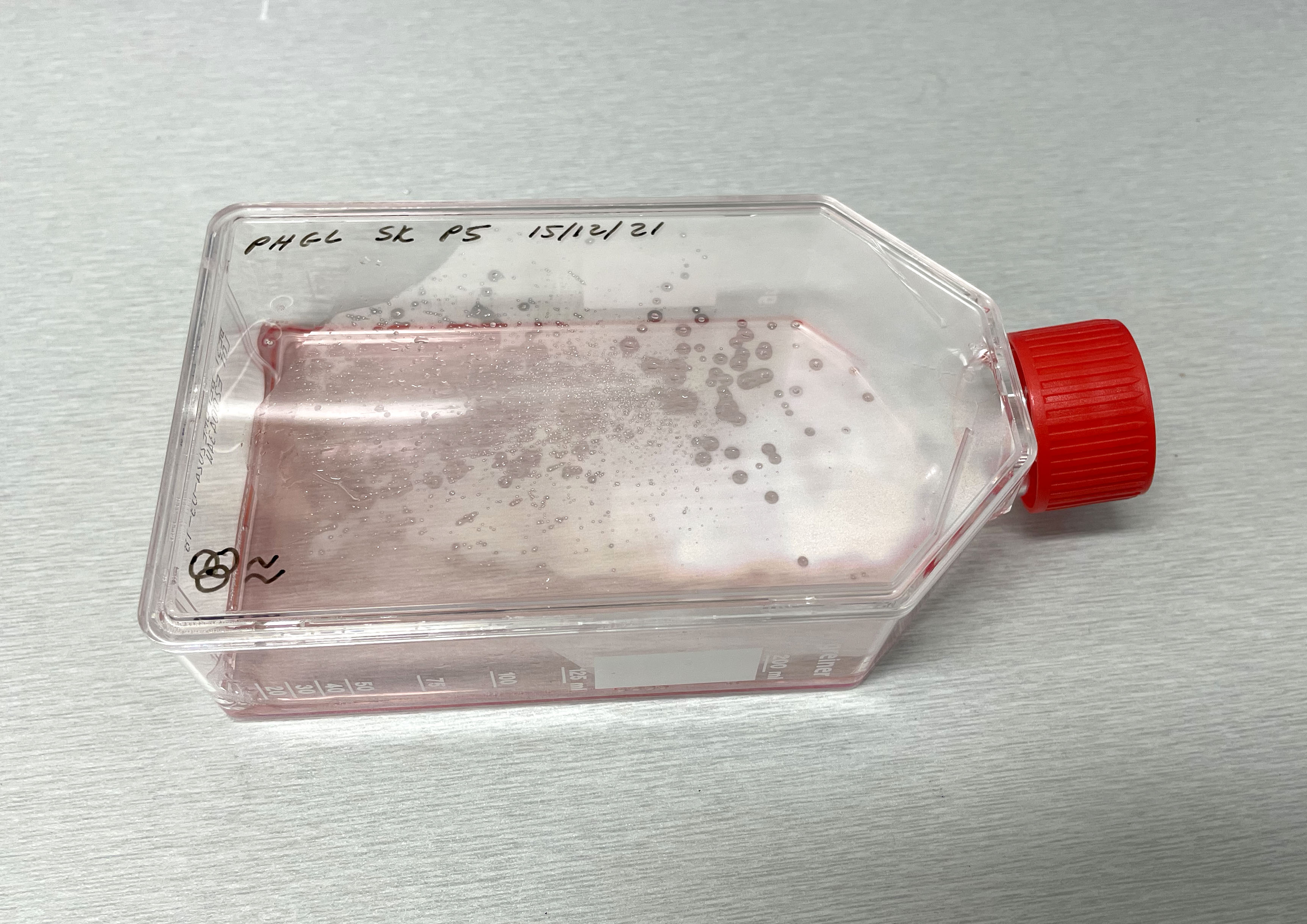 Photograph of PHGL Tumour Baby P5 in T75 flask plated on 15/12/21 prior to holidays.
Photograph of PHGL Tumour Baby P5 in T75 flask plated on 15/12/21 prior to holidays. 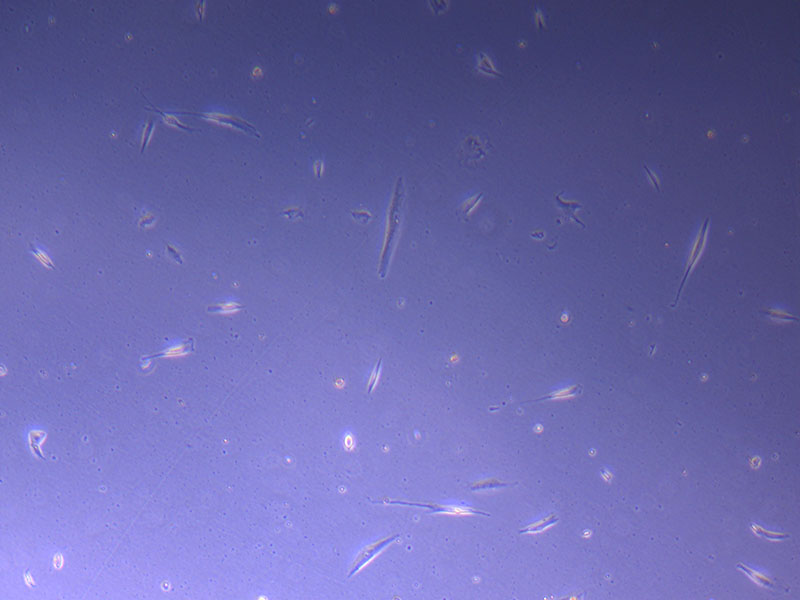
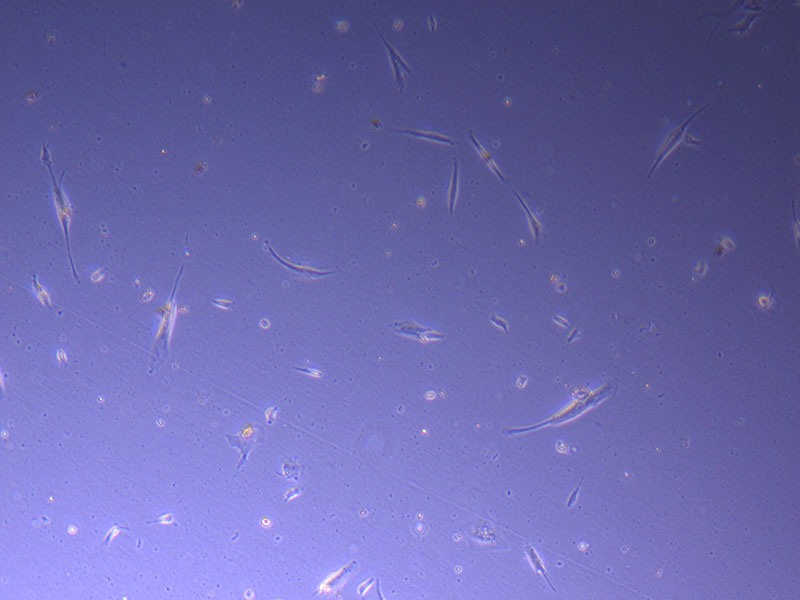 Light microscope image of PHGL Tumour Baby P5 in T75 flask plated on 15/12/21.
Light microscope image of PHGL Tumour Baby P5 in T75 flask plated on 15/12/21.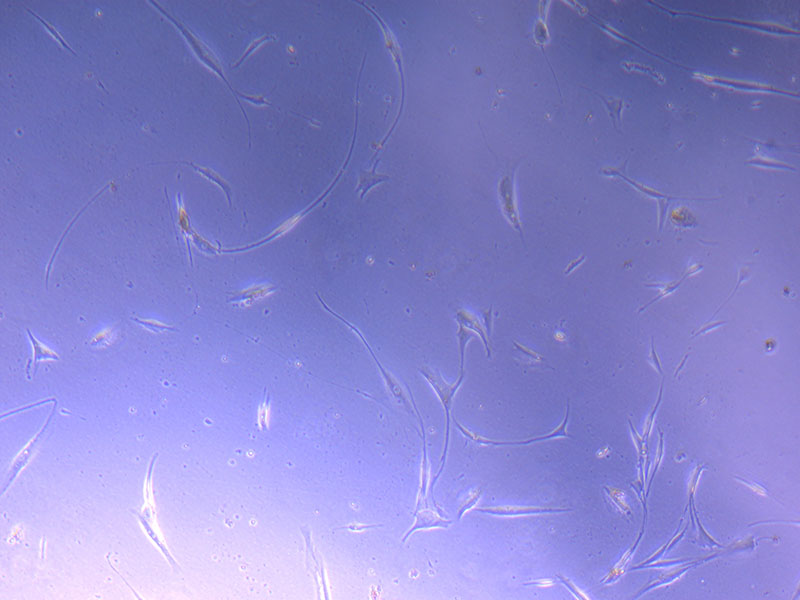
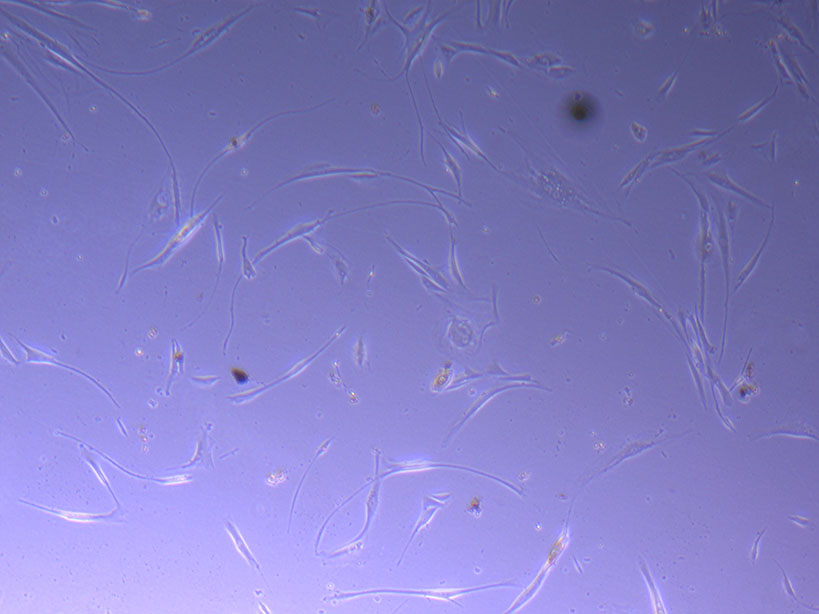 Light microscope images of PHGL Tumour Baby P5 in T75 flask plated on 15/12/21 and imaged on 13/1/22.
Light microscope images of PHGL Tumour Baby P5 in T75 flask plated on 15/12/21 and imaged on 13/1/22.  Photograph of PHGL Tumour Baby P5 in T75 flask #2 plated on 15/12/21.
Photograph of PHGL Tumour Baby P5 in T75 flask #2 plated on 15/12/21.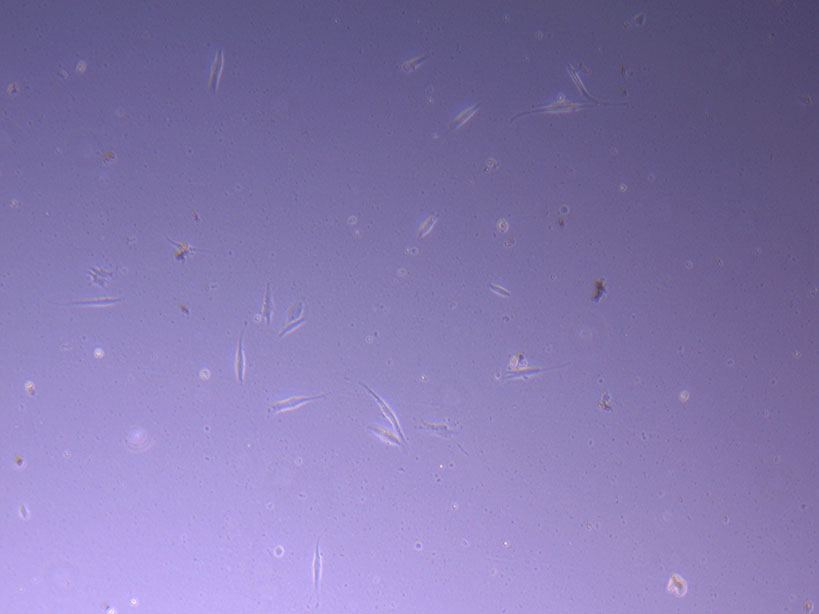 Light microscope image of PHGL Tumour Baby P5 in T75 flask 2 plated on 15/12/21.
Light microscope image of PHGL Tumour Baby P5 in T75 flask 2 plated on 15/12/21.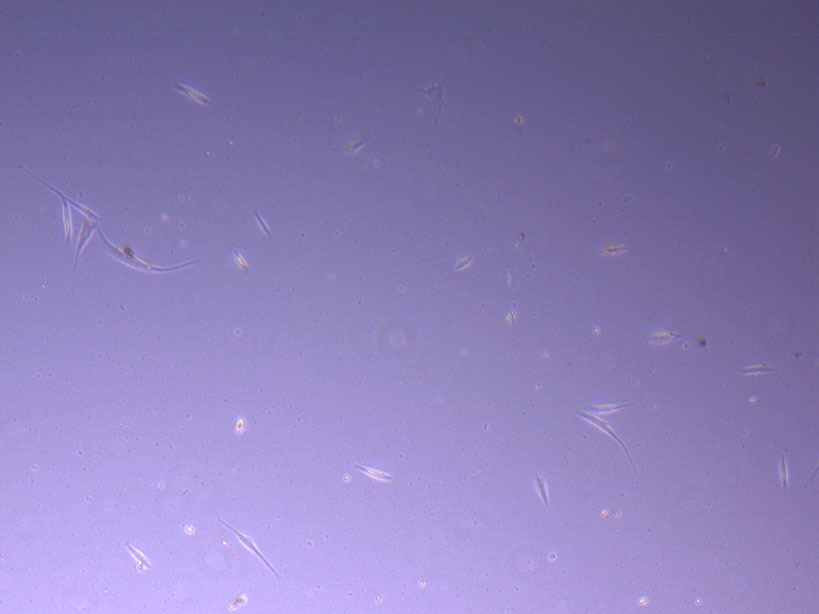 Light microscope image of PHGL Tumour Baby P5 in T75 flask 2 plated on 15/12/21.
Light microscope image of PHGL Tumour Baby P5 in T75 flask 2 plated on 15/12/21.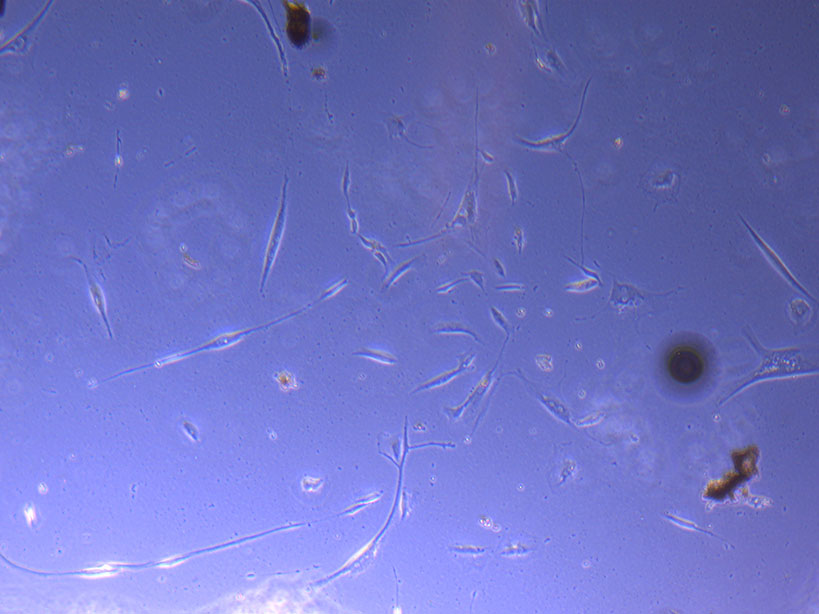
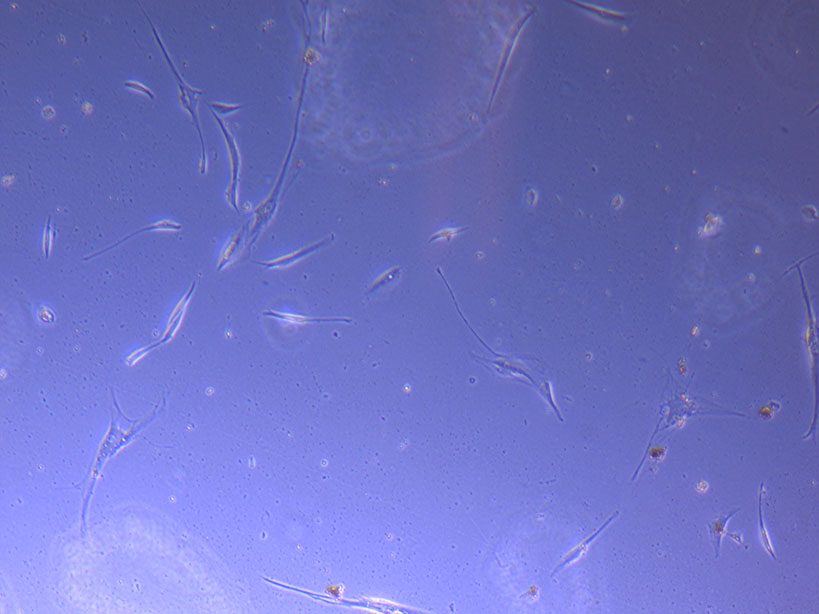 Light microscope image of PHGL Tumour Baby P5 in T75 flask 2 imaged on 13/1/22.
Light microscope image of PHGL Tumour Baby P5 in T75 flask 2 imaged on 13/1/22.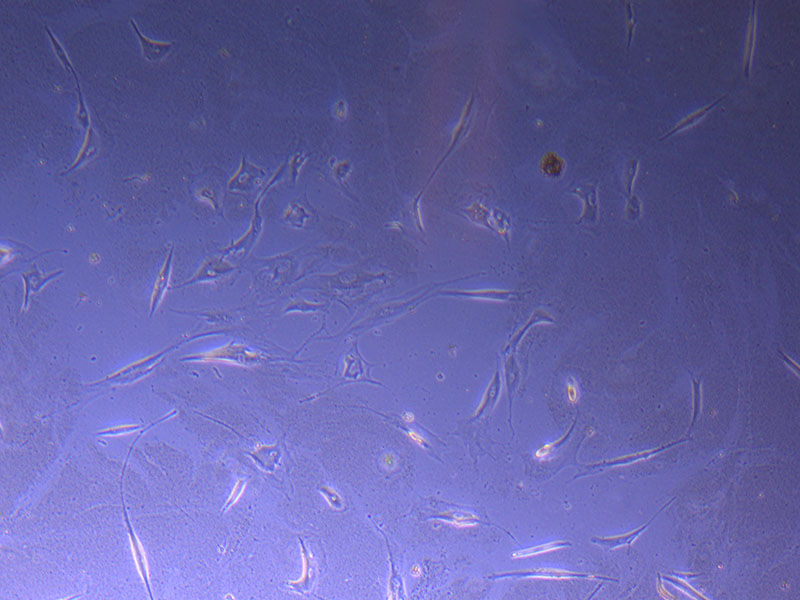 Light microscope image of PHGL TB cells at 13/1/22 showing a main cluster area of remaining cells.
Light microscope image of PHGL TB cells at 13/1/22 showing a main cluster area of remaining cells.  Photograph of P1 PHGL TB flask originally plated out by Jo-Maree on 10/0921.
Photograph of P1 PHGL TB flask originally plated out by Jo-Maree on 10/0921. 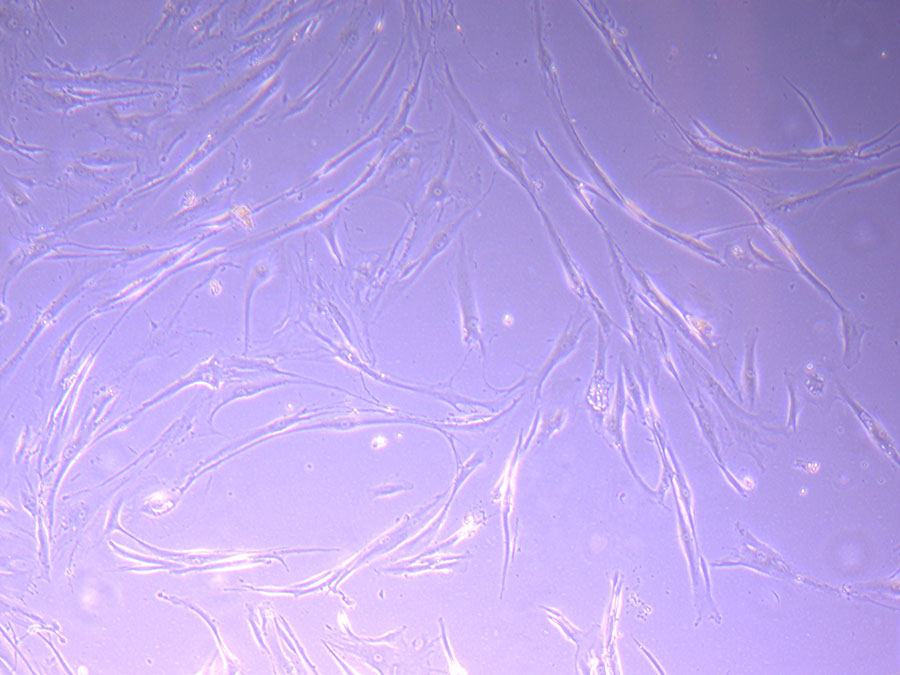 Light microscope image of P1 PHGL TB cells taken on 21/09/21 prior to passage.
Light microscope image of P1 PHGL TB cells taken on 21/09/21 prior to passage. 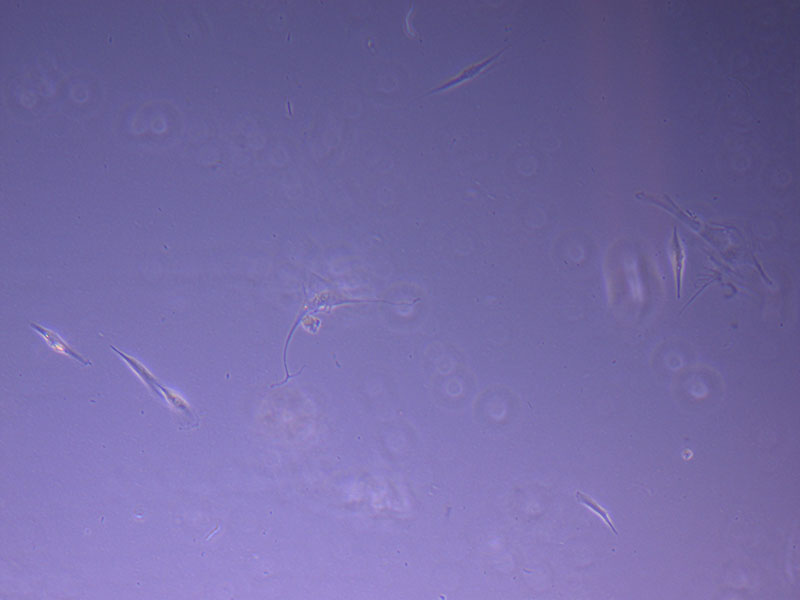 Light microscope image of P2 PHGL TB cells (in original flask) taken on 23/09/21 after cell passage on 21/09/21. A few cells remain visible in the flask.
Light microscope image of P2 PHGL TB cells (in original flask) taken on 23/09/21 after cell passage on 21/09/21. A few cells remain visible in the flask. 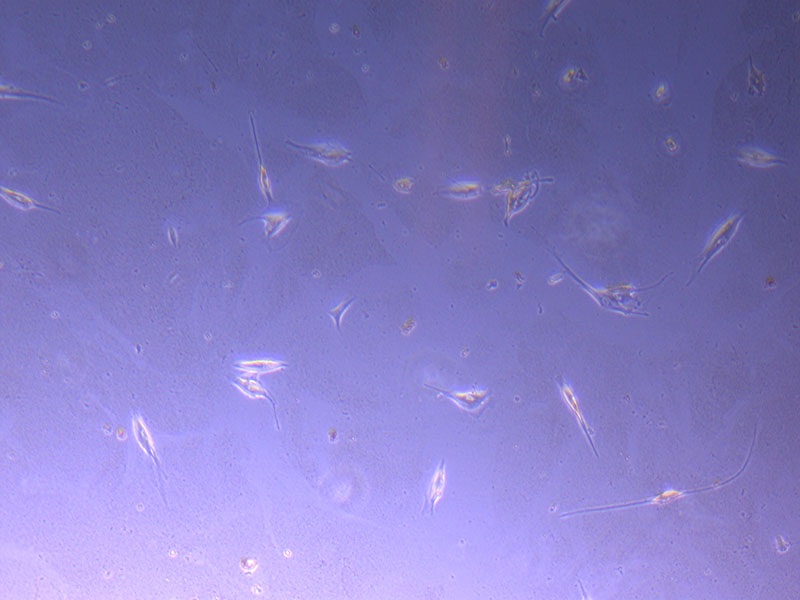 Light microscope image of P2 PHGL TB cells (in original flask) taken on 13/1/22. The image reveals a ‘sprinkle’ of cells beyond the main cluster.
Light microscope image of P2 PHGL TB cells (in original flask) taken on 13/1/22. The image reveals a ‘sprinkle’ of cells beyond the main cluster. 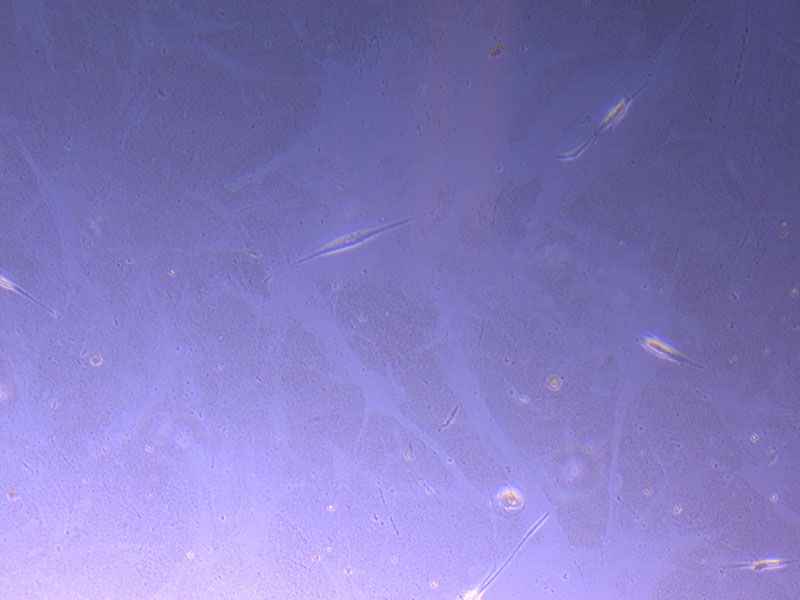
 Light microscope images of P2 PHGL TB cells (in original flask) taken on 13/1/22. The image reveals trails of cellular movement and existence.
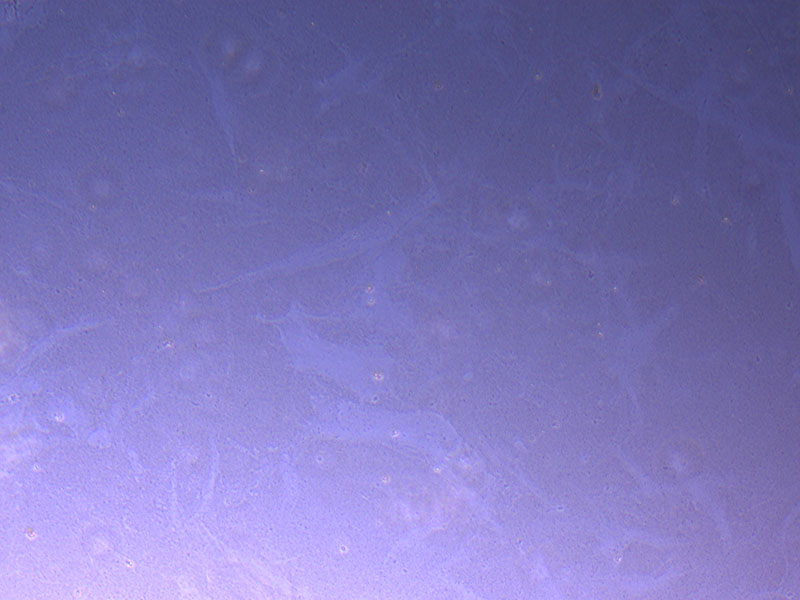
Light microscope images of P2 PHGL TB cells (in original flask) taken on 13/1/22. The image reveals trails of cellular movement and existence.  Light microscope image of cut glass dish with media containing dead cell debris and evidence of a small number of surviving cells.
Light microscope image of cut glass dish with media containing dead cell debris and evidence of a small number of surviving cells. 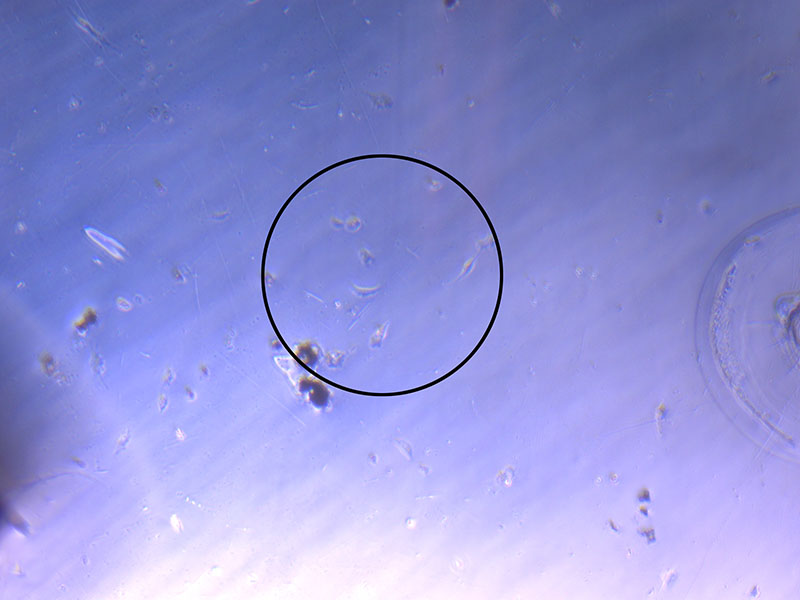
 Light microscope image of cut glass dish in PBS showing further evidence of a small number of surviving cells.
Light microscope image of cut glass dish in PBS showing further evidence of a small number of surviving cells.  Light microscope image of cut glass dish in PBS with likely cells circled. There are a few additional potential cells visible, but I have only circled the most obvious.
Light microscope image of cut glass dish in PBS with likely cells circled. There are a few additional potential cells visible, but I have only circled the most obvious. 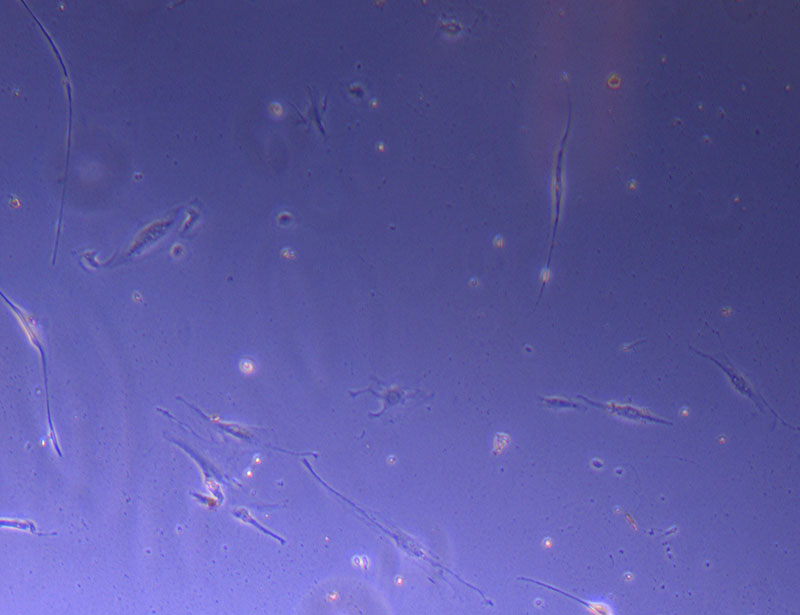 Light microscope image of Petri dish with fresh complete media with live cells.
Light microscope image of Petri dish with fresh complete media with live cells. 
 Simple graphic interface with presets ready to complete fluorescent microscopy.
Simple graphic interface with presets ready to complete fluorescent microscopy. 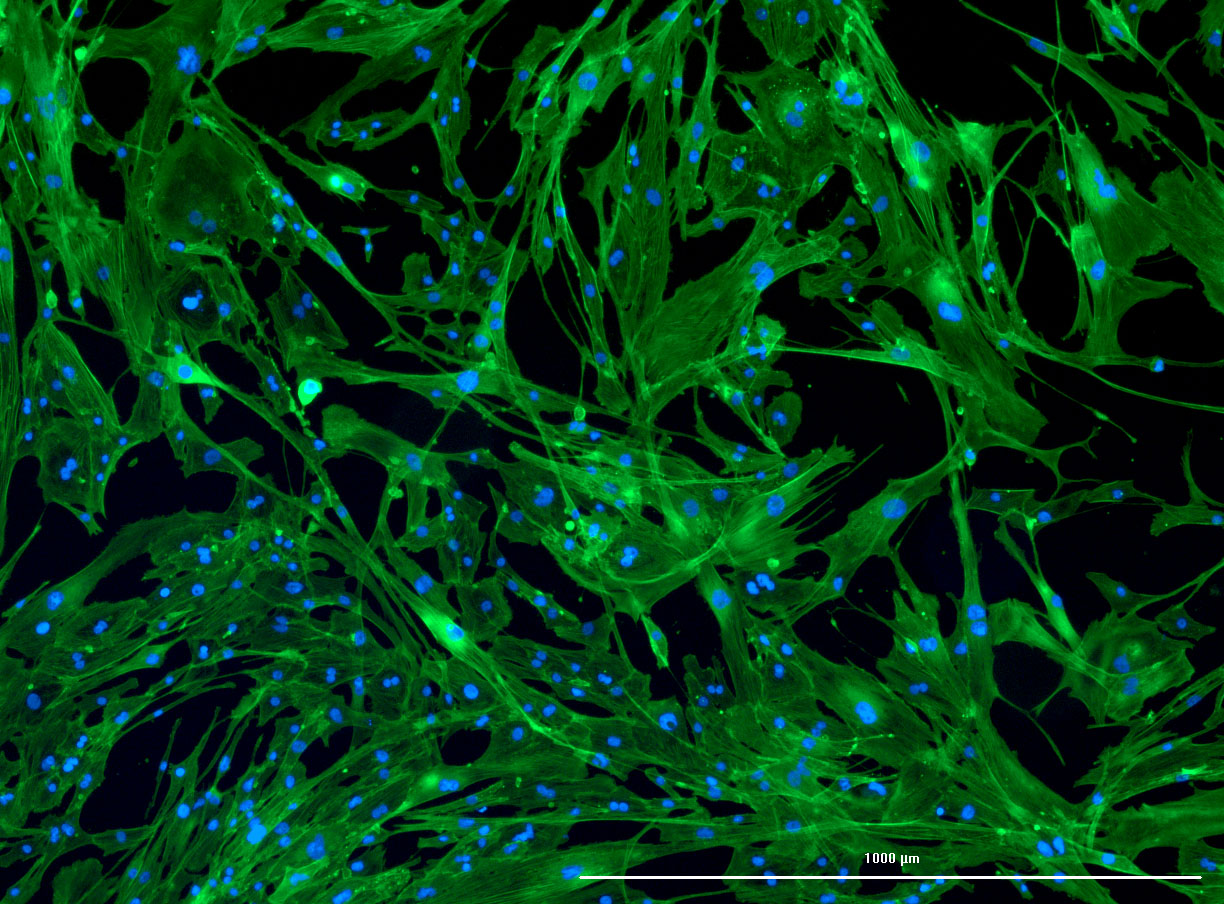
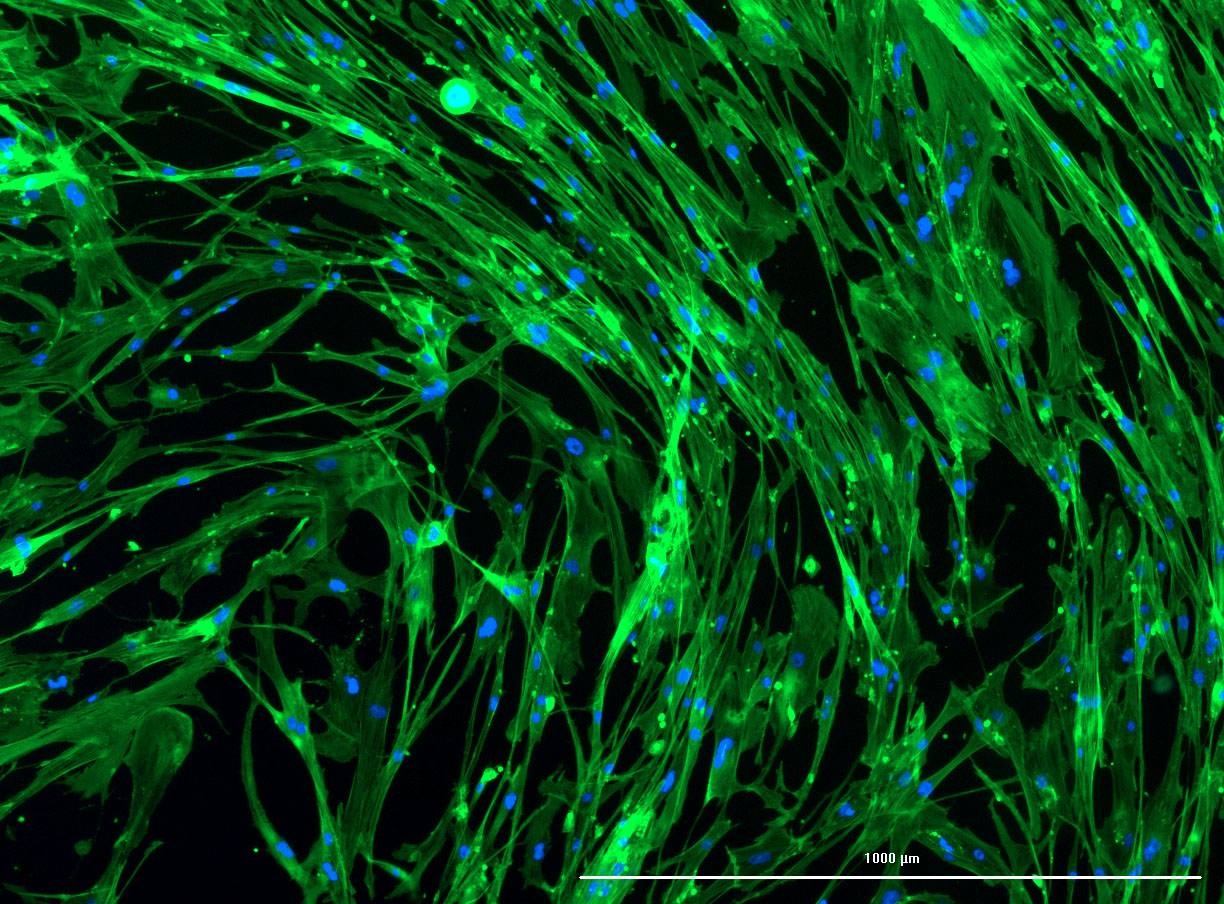
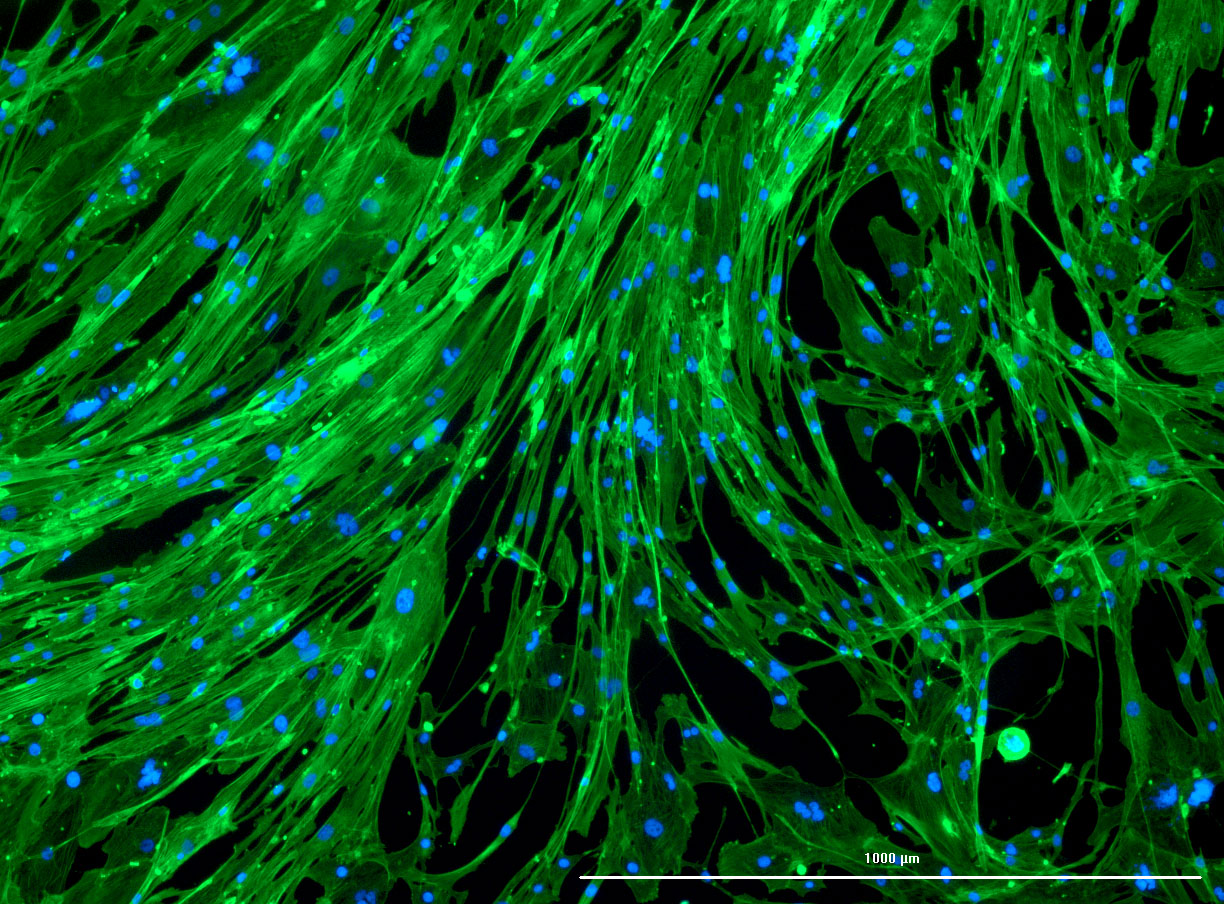
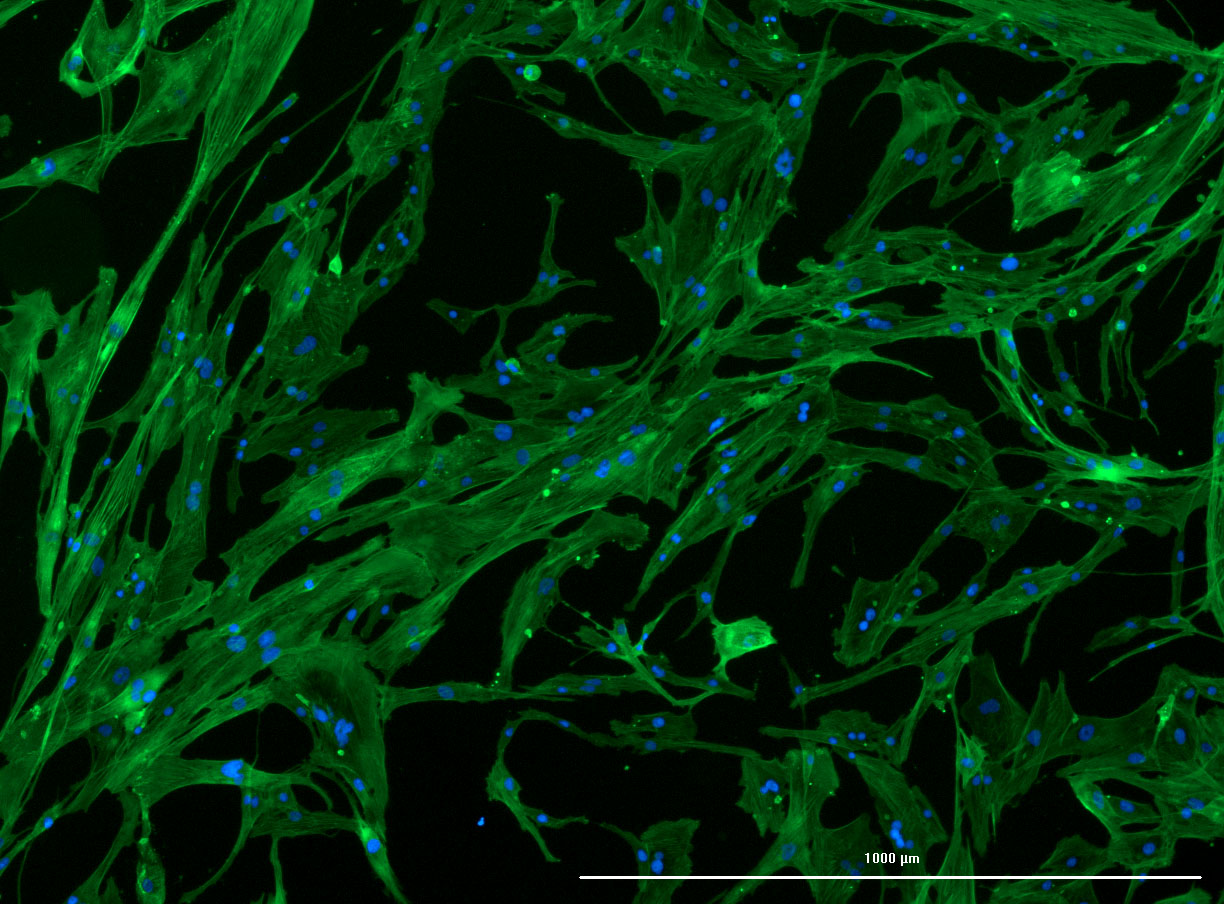 DAPI and Fluorescein Phalloidin staining of confluent fibroid cells P4 (although this is potentially misleading as the cells are very slow growing).
DAPI and Fluorescein Phalloidin staining of confluent fibroid cells P4 (although this is potentially misleading as the cells are very slow growing). 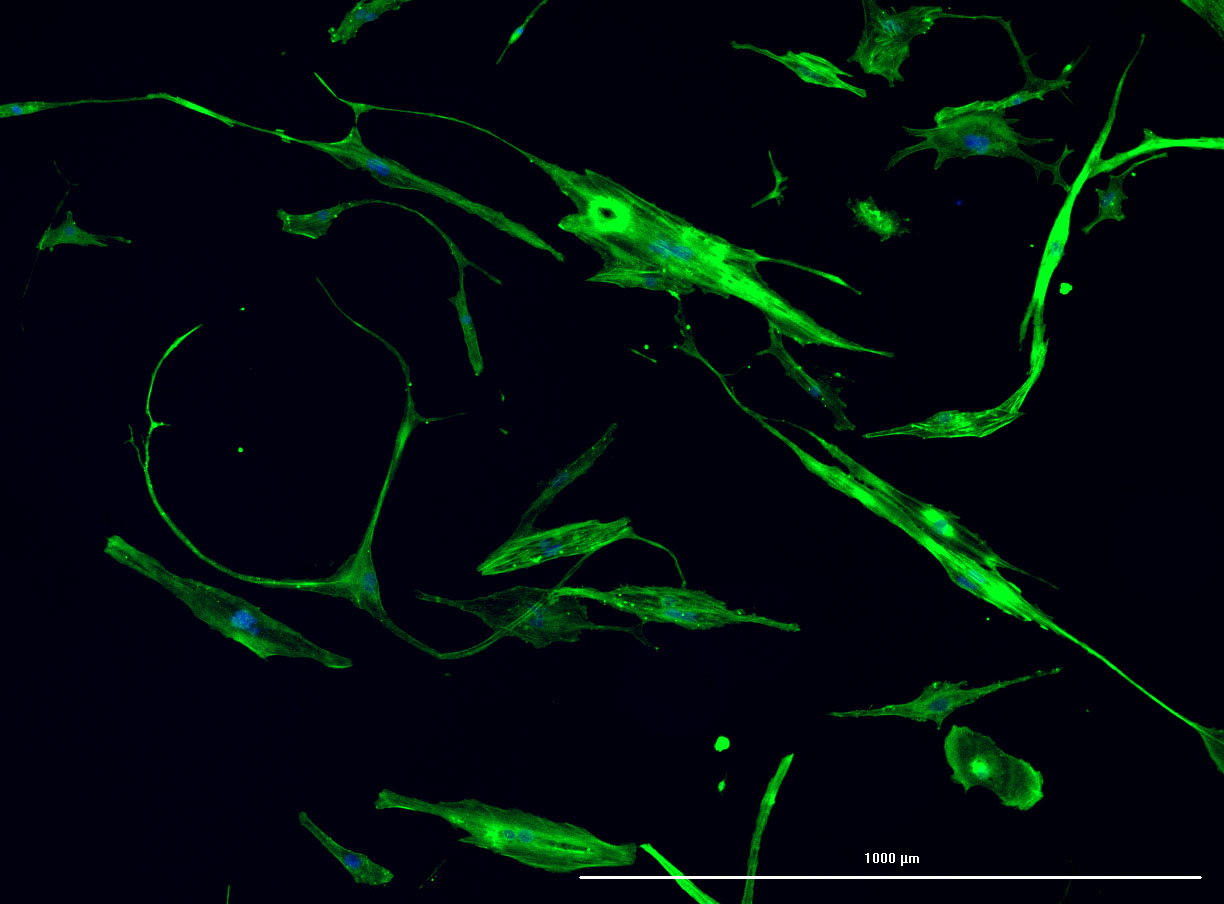
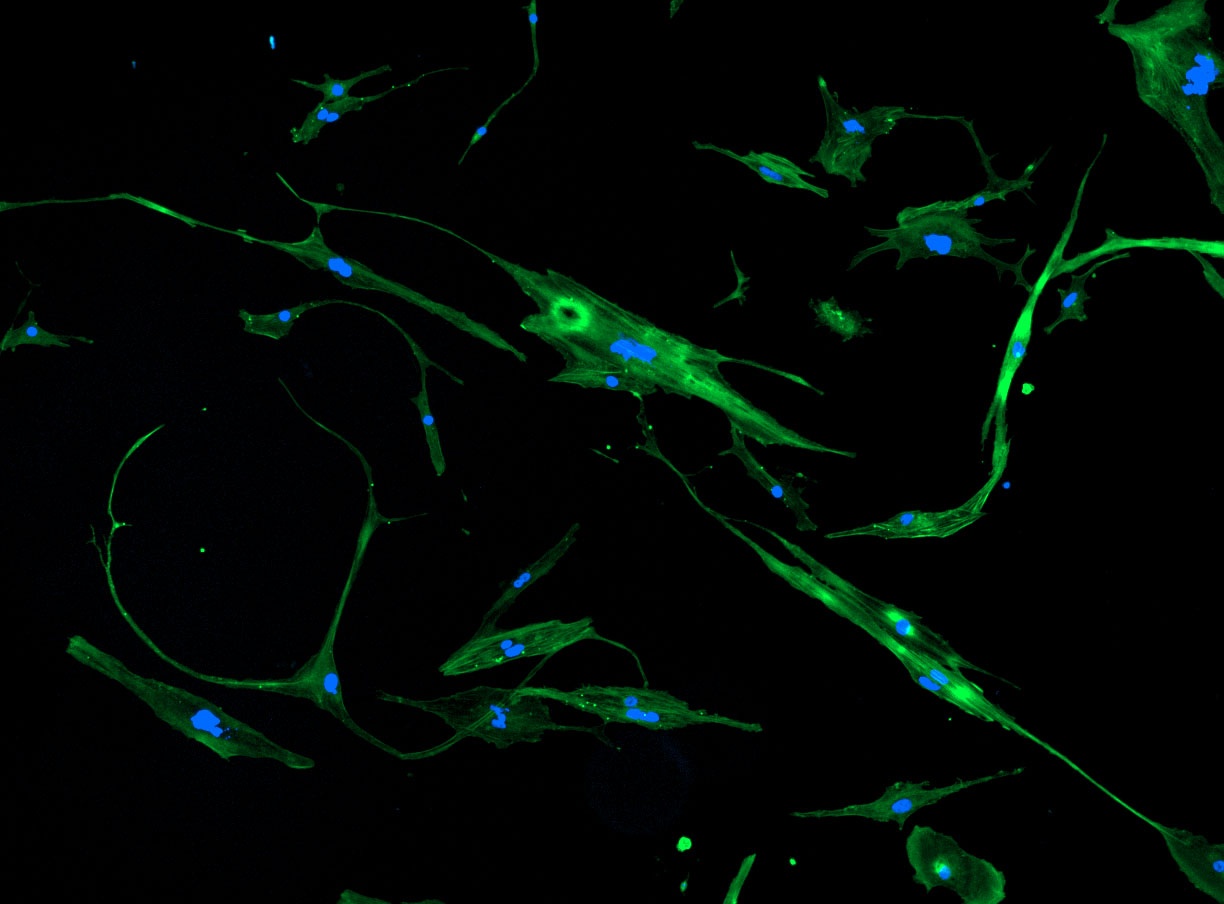
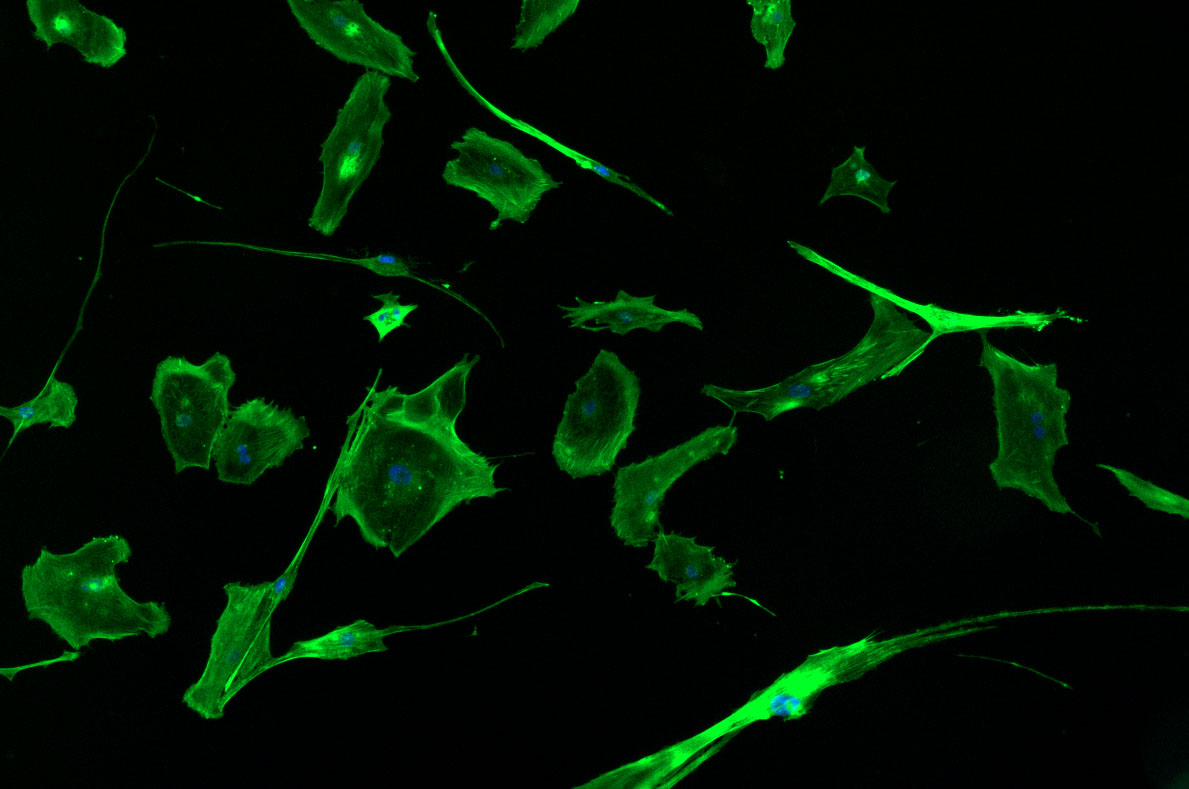
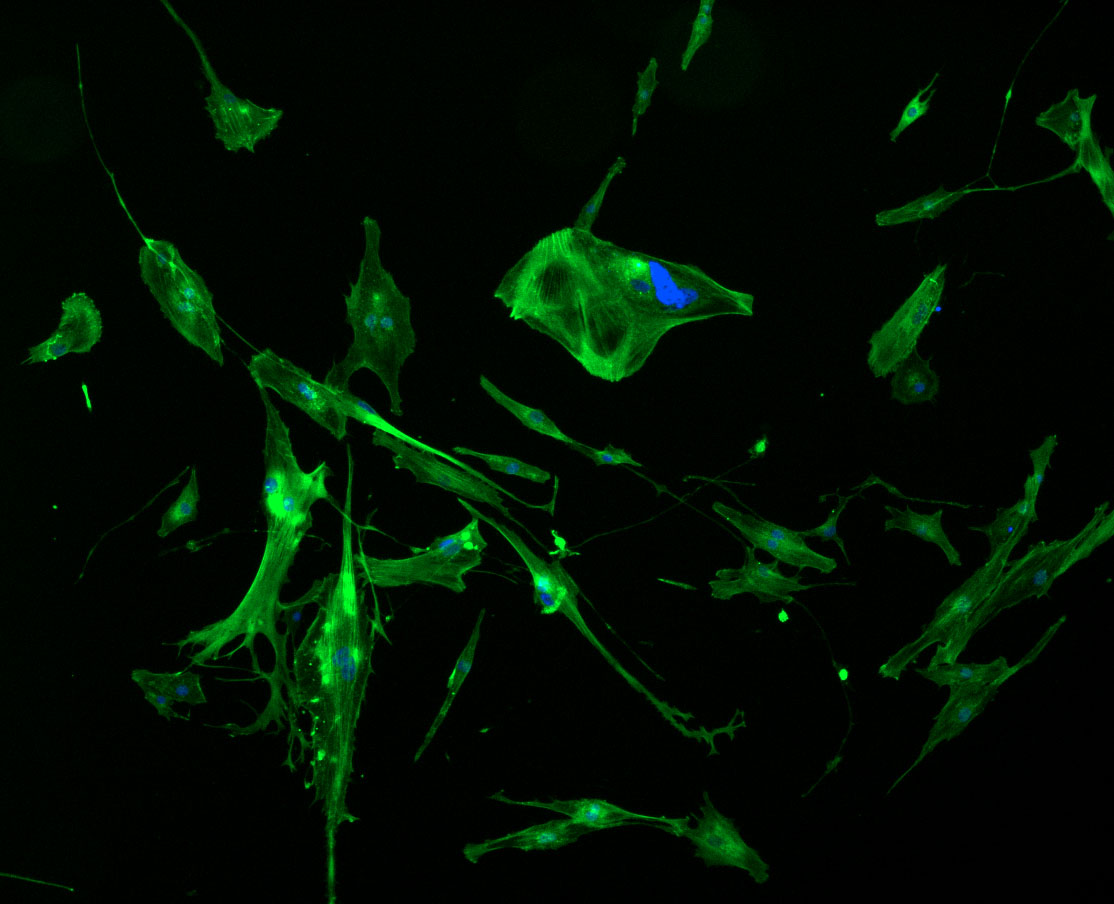
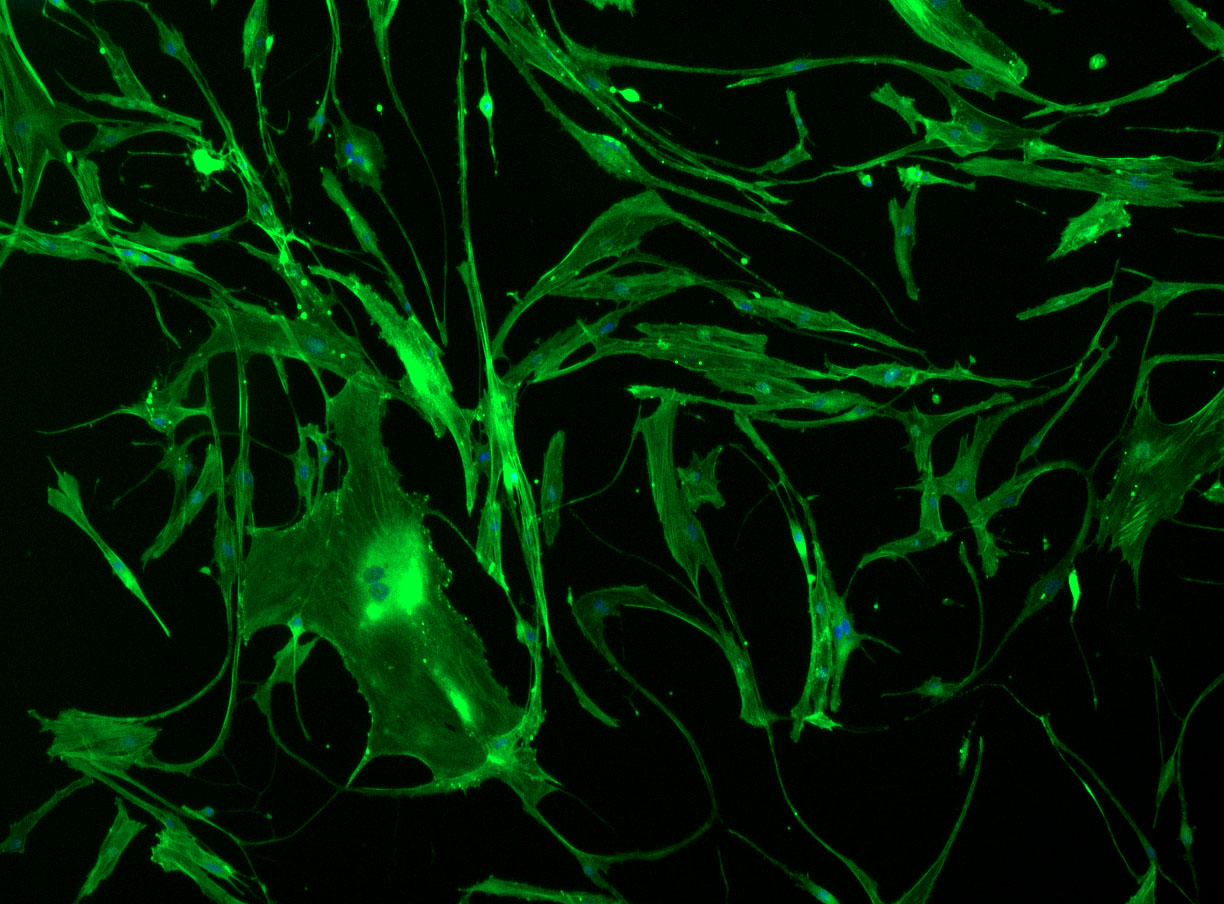
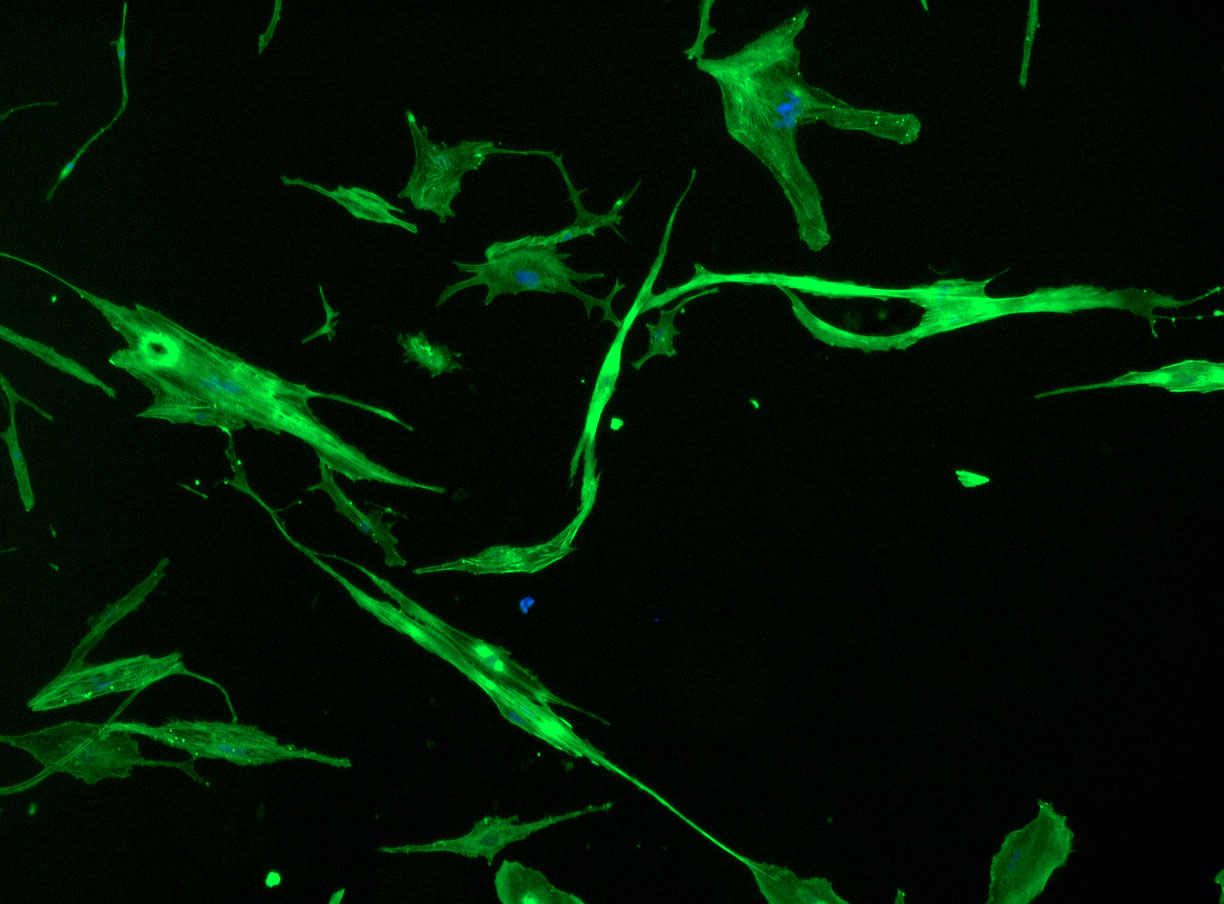 DAPI and Fluorescein Phalloidin staining of fibroid cells P4 which enables better visualisation of individual cells.
DAPI and Fluorescein Phalloidin staining of fibroid cells P4 which enables better visualisation of individual cells.  Working with 4% PFA in Fume Hood
Working with 4% PFA in Fume Hood Working at lab bench in the Stroke Group area
Working at lab bench in the Stroke Group area

 Unwrapping Petri dishes and getting ready to coat culture glassware with PLL.
Unwrapping Petri dishes and getting ready to coat culture glassware with PLL.  PLL coated glassware in Petri dishes ready for incubation.
PLL coated glassware in Petri dishes ready for incubation.  Cut glass vessels with cells ready for incubation.
Cut glass vessels with cells ready for incubation. 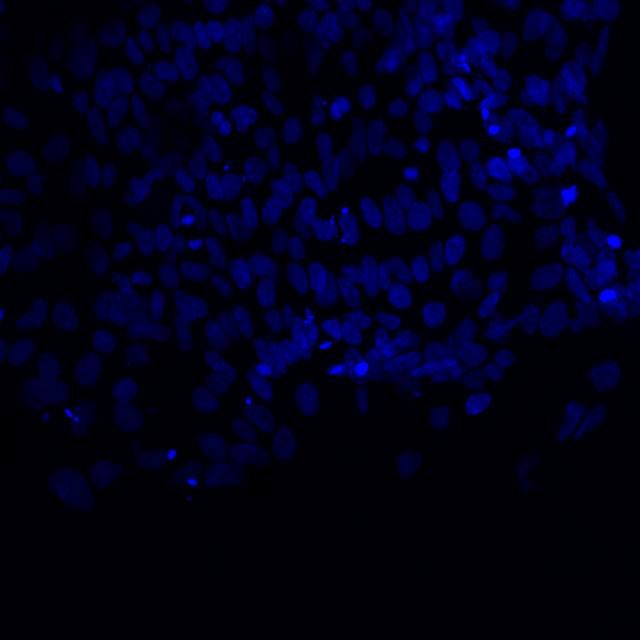 Microscopic image of stem cells, Hues 9 stained with DAPI (blue) by the UC San Diego Stem Cell Program.
Microscopic image of stem cells, Hues 9 stained with DAPI (blue) by the UC San Diego Stem Cell Program. Image of Amanita phalloides via
Image of Amanita phalloides via 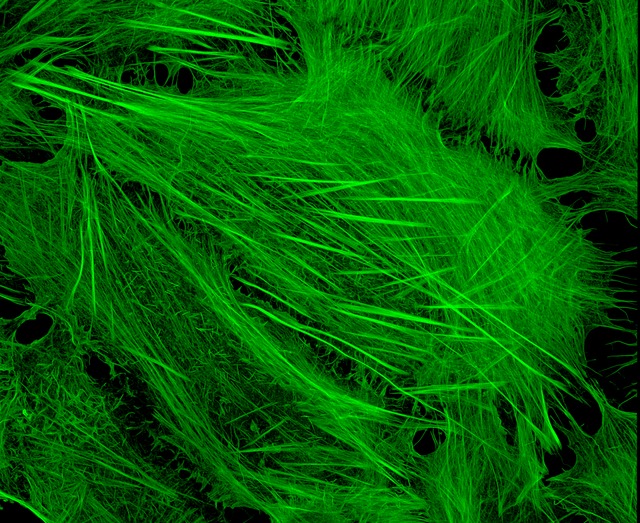 U2OS cells stained with fluorescent phalloidin taken on a confocal microscope by Howard Vindin
U2OS cells stained with fluorescent phalloidin taken on a confocal microscope by Howard Vindin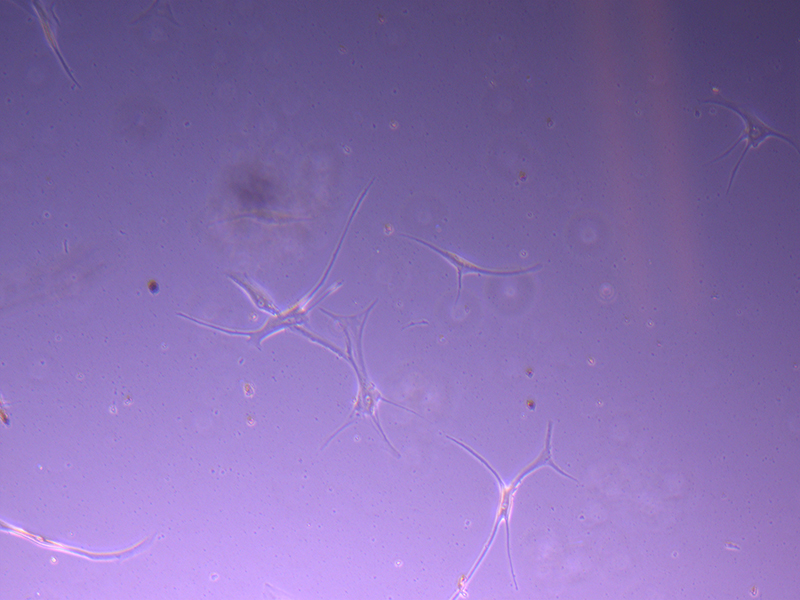 T25 – Flask 1 P 3, 11/10/21
T25 – Flask 1 P 3, 11/10/21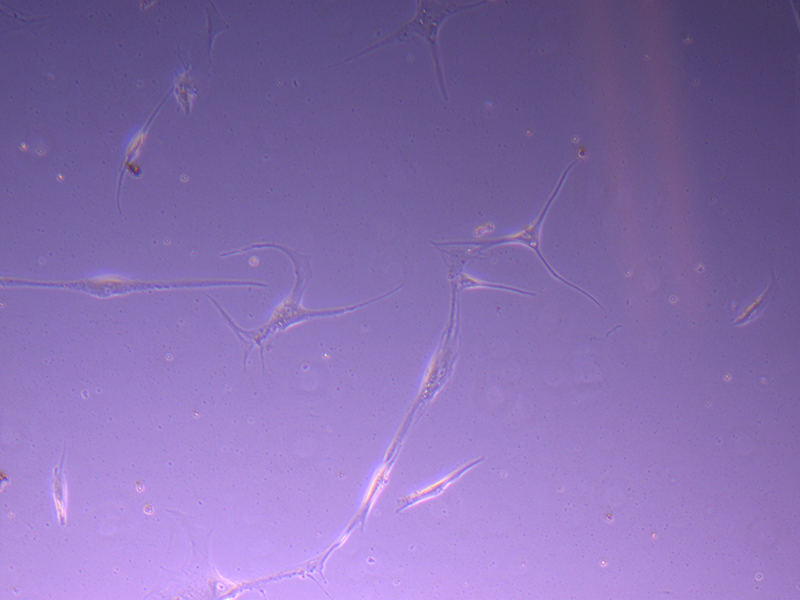 T25 – Flask 1 P 3, 11/10/21
T25 – Flask 1 P 3, 11/10/21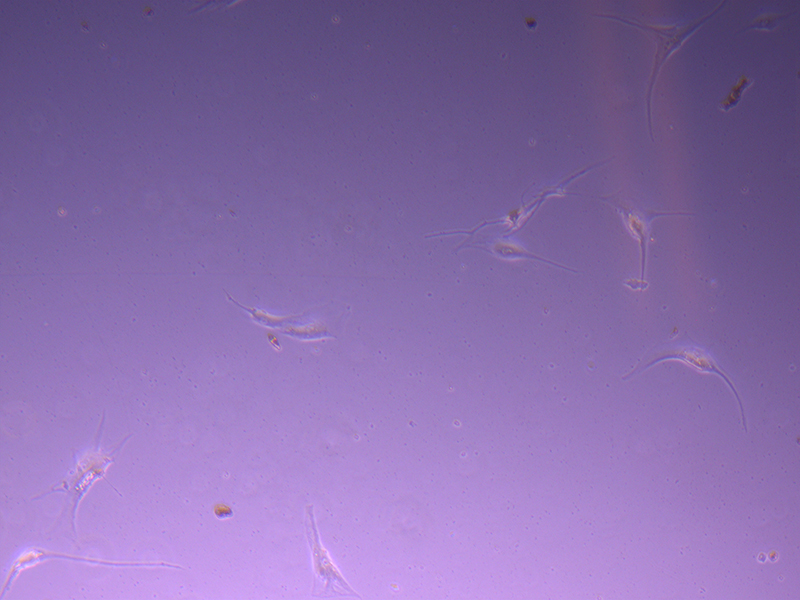 T25 – Flask 2 P 3, 11/10/21
T25 – Flask 2 P 3, 11/10/21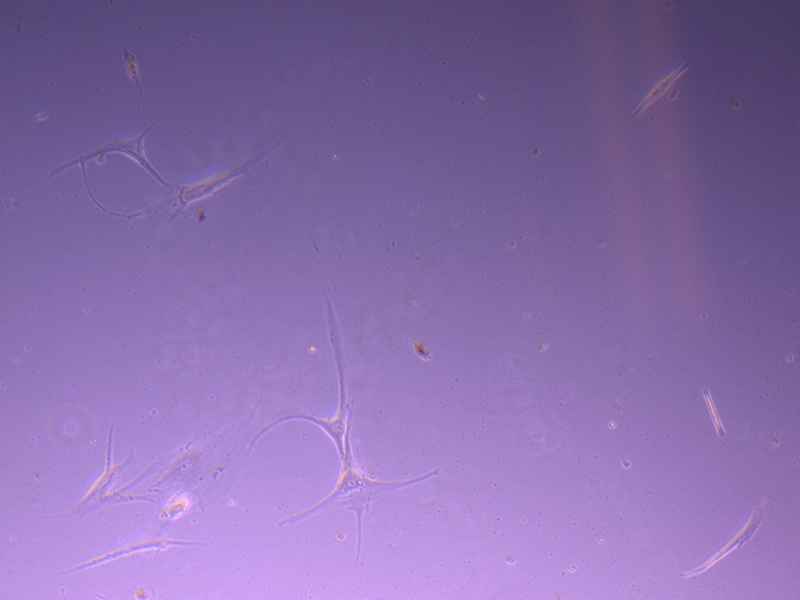 T25 – Flask 2 P 3, 11/10/21
T25 – Flask 2 P 3, 11/10/21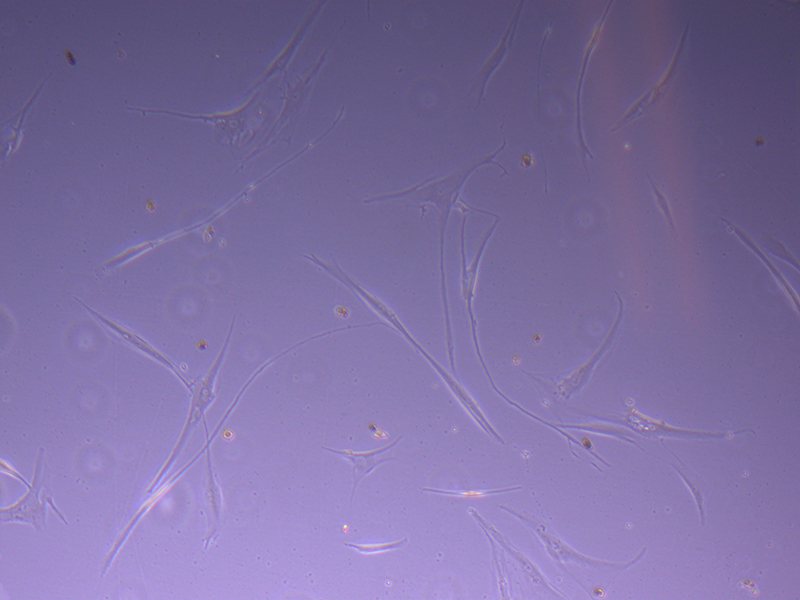 T75 – Flask 1 P 3, 11/10/21
T75 – Flask 1 P 3, 11/10/21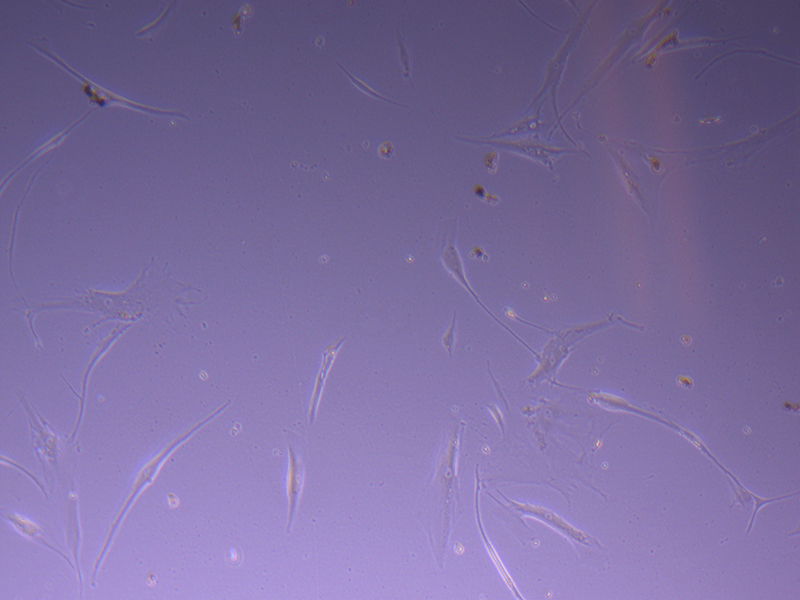 T75 – Flask 1 P 3, 11/10/21
T75 – Flask 1 P 3, 11/10/21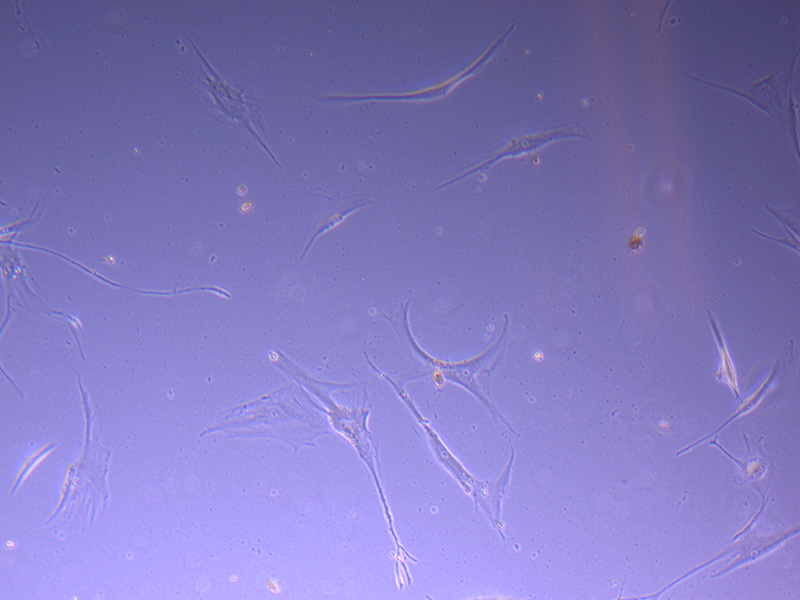 T75 – Flask 2 P 3, 11/10/21
T75 – Flask 2 P 3, 11/10/21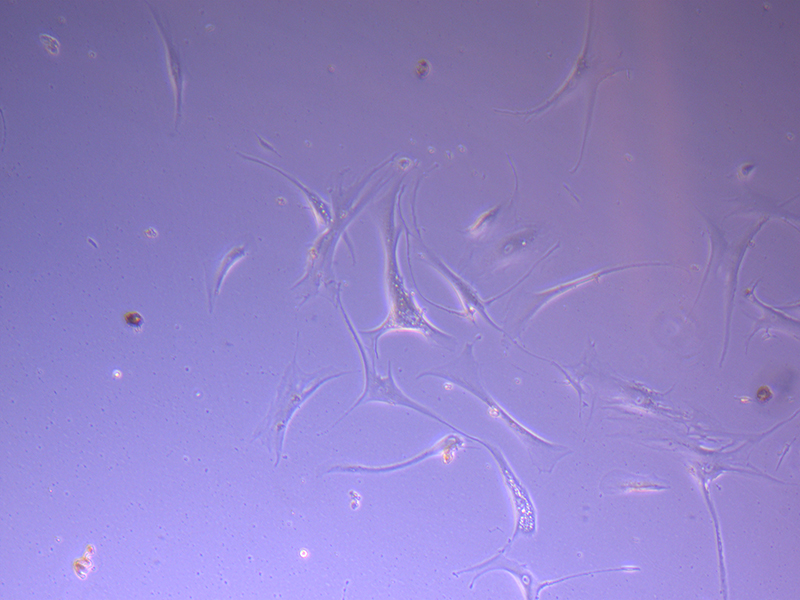 T75 – Flask 2 P 3, 11/10/21
T75 – Flask 2 P 3, 11/10/21 Images of PHGL Tumour Baby Cells P2 at 1/10/21
Images of PHGL Tumour Baby Cells P2 at 1/10/21{kind=link}