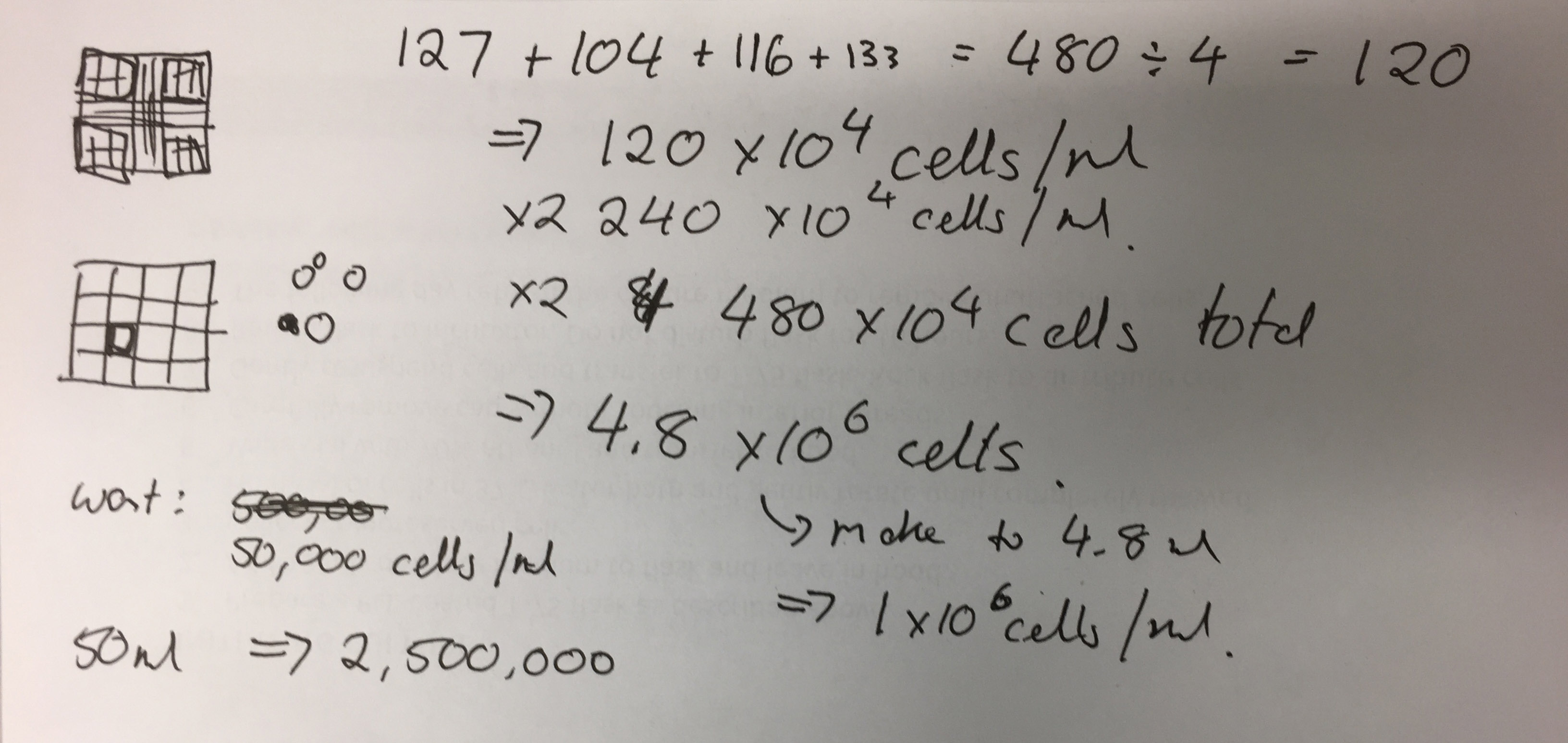We had a detailed project planning and ethics clearance review meeting on Monday. It was heartening to hear that the ethics clearance documentation is almost ready for formal submission.
These are some of the processes we are expecting to undertake:
- Cell culture of fibroid cells (optimisation of culture methods for 2D and 3D environments, cellular response and proliferation testing)
- Co-culture of fibroid cells with other cell lines
- Genetic profiling of primary fibroid cells
- Immortalisation of primary fibroid cells via established commercial kit (e.g. Applied Biological Materials (ABM) cell immortalisation kits)
- Reprogramming of primary cells to generate induced pluripotent stem cells (iPSCs) via established commercial kit (e.g. Epi5™ Episomal iPSC Reprogramming Kit available via Thermo Fisher)
- Cell culture of reprogrammed or immortalised fibroid cells (optimisation of culture methods for 2D and 3D environments, cellular response and proliferation testing)
- Genetic profiling of reprogrammed or immortalised fibroid cells and cells lines (if successful)
- Fixing and staining of cells
- Light and confocal microscopy of cultured cells
- Timelapse video of cultured cells
Other potential processes include:
- Green Fluorescent Protein (GFP) cell tagging [UTAS]
- Scanning Electron Microscopy (SEM) of cultured cells [UTAS]
- Transmission Electron Microscopy (TEM) of cultured cells [UTAS]
- Histopathology of cultured 3D structures [UTAS or QUT]
- Development of Gastruloids, Organoids or Neurospheres (self-organised 3D cell masses) [UTAS]
I was delighted to hear that my desires for creating aggregates of cells via the production of gastruloids or organoids was not outside the domain of possibility. Jo-Maree has produced neurospheres (balls of neural stem cells) previously, so there may be scope (and hopefully time) to experiment with cell clusters.
Gastuloids are of particular interest to me as they are cell clusters that display features of early embryo development. Staining and fluorescence imaging (apart from being visually stunning) enables the visualisation of tissue organisation as shown in this figure and caption from the Nature publication ‘Multi-axial self-organization properties of mouse embryonic stem cells into gastruloids’:
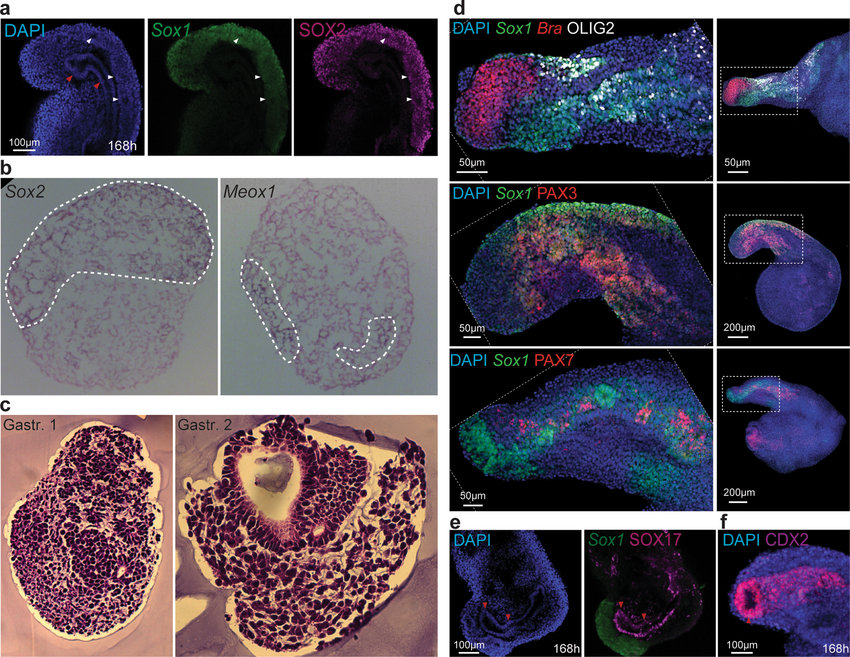
Tissue organization in gastruloids a, Gastruloids formed from Sox1GFP;BramCherry (SBR) line and stained for Sox2 expression (Sox1GFP and SOX2 signals are displayed in green and magenta, respectively). White arrowheads indicate tubular SOX2/Sox1-positive neural structures. Red arrowheads point to the presumptive digestive tube. b, WISH on 8-µm transverse cryosections of gastruloids at 144 h AA using Sox2 and Meox1 antisense probes, counter-stained with Nuclear Fast Red. Sox2-positive cells localized predominantly in a compact dorsal domain, whereas Meox1 signals were found in two bilateral domains. The domain of expression of each gene is outlined with white dashed lines. c, Haematoxylin and eosin staining of transverse paraffin sections of different gastruloids at 120 h AA, showing the diversity of cell types and several levels of tissue organization. d, Gastruloids formed from Sox1GFP;BramCherry ESCs were fixed and stained at 168 h AA for OLIG2 (top, white), PAX3 (middle, red) and PAX7 (bottom, red). Scale bars as indicated. c, d, Gastruloids formed from Sox1GFP; BramCherry ESCs collected at 168 h AA and stained for SOX17 (magenta, c) or CDX2 (magenta, d). Scale bars as indicated.
Gastruloids are created from embryonic stem cells, although iPSC cells (cells that have been reprogrammed into a stem cell like state) have also been used. As such, if I have success with reprogramming my fibroid cells to iPSC cells, I could use them to make, and learn more about, gastruloids.
During our discussion, we decided it would be a good idea to include an optional alternative to the fibroid cells – just in case there is an issue with contamination or the freezing/thawing process. While I have isolated skin cells (fibroblasts) from hair follicles and skin grafts previously as part of the HSE Project at QUT, we decided on skin scrapings. This approach was selected as will enable the isolation of skin cells, is not too invasive and is well established within the School of Medicine.
Once I’ve included this information including the protocol, I should be ready to submit the final ethics clearance document.
Beccari, L., Moris, N., Girgin, M., Turner, D.A., Baillie-Johnson, P., Cossy, A.C., Lutolf, M.P., Duboule, D. and Arias, A.M., 2018. Multi-axial self-organization properties of mouse embryonic stem cells into gastruloids. Nature, 562(7726), pp.272-276.